




Содержание
Перейти к:
https://doi.org/10.14341/DM12648
Перейти к:
Сердечная недостаточность (СН) является актуальной проблемой общественного здравоохранения. Согласно литературным данным, наличие сахарного диабета (СД) значительно увеличивает риск повторных госпитализаций и длительность пребывания в стационаре у пациентов с СН. Доля СН остается высокой вследствие увеличения продолжительности жизни, большей распространенности факторов риска и улучшения показателей выживаемости. В настоящее время достижения в лечении ишемической болезни сердца (ИБС) и патологии клапанного аппарата значительно улучшили показатели выживаемости, однако прогноз при СН остается крайне неблагоприятным. Среди наиболее важных медицинских проблем особое место занимает СН у пациентов с СД 2 типа (СД2). СД2 способствует возникновению СН с помощью различных механизмов, включая комплекс специфических структурных, функциональных и метаболических изменений со стороны миокарда, называемых диабетической кардиомиопатией. Несмотря на активное изучение причин кардиомиопатии, поиск и внедрение новых подходов в оценке риска развития данного патологического феномена у пациентов с СН остаются актуальными. В данном обзоре рассматриваются современные гипотезы развития диабетической кардиомиопатии, такие как инсулинорезистентность, эндотелиальная дисфункция, фиброз, липотоксичность и энергетические нарушения.
Сваровская А.В., Гарганеева А.А. Сахарный диабет 2 типа и сердечная недостаточность — современный взгляд на механизмы развития. Сахарный диабет. 2022;25(3):267-274. https://doi.org/10.14341/DM12648
Svarovskaya A.V., Garganeeva A.A. Diabetes mellitus and heart failure — a modern look at the mechanisms of development. Diabetes mellitus. 2022;25(3):267-274. (In Russ.) https://doi.org/10.14341/DM12648
Сердечная недостаточность (СН) является актуальной проблемой общественного здравоохранения. Согласно литературным данным, наличие сахарного диабета (СД) значительно увеличивает риск повторных госпитализаций и длительность пребывания в стационаре у пациентов с СН [1]. Доля СН остается высокой вследствие увеличения продолжительности жизни, большей распространенности факторов риска и улучшения показателей выживаемости [2][3]. В настоящее время достижения в лечении ишемической болезни сердца (ИБС) и патологии клапанного аппарата значительно улучшили показатели выживаемости, однако прогноз при СН остается крайне неблагоприятным [4].
Среди наиболее важных медицинских проблем особое место занимает СН у пациентов с СД 2 типа (СД2). Согласно данным регистра REACH, у пациентов с СД2 СН увеличивает риск сердечно-сосудистой смерти примерно на 250%, количество госпитализаций по поводу СН — на 500% [5]. Кроме того, СН у пациентов с диабетом ассоциируется со средней продолжительностью жизни около 4 лет от момента постановки диагноза [6]. СД может способствовать возникновению СН с помощью различных механизмов, включающих комплекс специфических структурных, функциональных и метаболических изменений со стороны миокарда, называемых диабетической кардиомиопатией [7].
Несмотря на активное изучение причин кардиомиопатии, поиск и внедрение новых подходов в оценке риска развития данного патологического феномена у пациентов с СН остаются актуальными.
Патогенез СН при СД2 практически всегда складывается из метаболического и ишемического компонентов. Взаимосвязь сердечно-сосудистых заболеваний (ССЗ) и СД необходимо рассматривать в рамках единой концепции кардио-рено-церебрально-метаболического или кардио-рено-церебрально-диабетического континуума, поскольку она носит непрерывный характер и обусловлена общими факторами риска и механизмами прогрессирования [8].
Формирование СН у больных СД2 обусловлено сочетанием ИБС с диабетической микроангиопатией, снижающей резерв коронарного кровотока, диастолической дисфункцией левого желудочка (ЛЖ). Одним из осложнений СД2, оказывающим существенное воздействие на клиническую картину и прогноз ССЗ, является диабетическая кардиальная автономная нейропатия (ДКАН). Наличие ДКАН отмечается у больных с длительным течением СД2, которое ухудшает качество жизни пациентов и сочетается с высокой летальностью [9]. Метаанализ 15 продольных исследований [10], которые включали в общей сложности 2900 пациентов, наблюдавшихся в течение 1–16 лет, показал, что наличие ДКАН обусловливает более высокий риск смертности. Наиболее опасным проявлением ДКАН является бессимптомная ишемия миокарда.
Формирование ДКАН происходит за счет трех основных патофизиологических механизмов:
-ускоренного атеросклеротического поражения коронарных артерий (вследствие усиления пролиферации гладкомышечных клеток сосудистой стенки, стимулирования процессов воспаления, тромбообразования, эндотелиальной дисфункции, дислипидемии) с повышенным риском ишемии миокарда (из-за повышенной уязвимости атеросклеротических бляшек), развитием инфаркта, постинфарктного ремоделирования ЛЖ с последующим каскадом нарушений, приводящих к развитию ишемической кардиомиопатии;
-повышенной предрасположенности к развитию гипертрофии ЛЖ и усилению процессов фиброзирования миокарда, что способствует увеличению жесткости миокарда, нарушению процессов расслабления и нарастанию диастолических нарушений ЛЖ;
-создание условий для нарушения энергетического баланса миокардиоцитов (МКЦ) вследствие дефектов утилизации глюкозы и свободных жирных кислот (СЖК) с аккумуляцией липидов в МКЦ, формированием липотоксичности, усилением апоптоза МКЦ и, в конечном итоге, развитием систолической дисфункции ЛЖ [11].
Термины «ишемическая кардиомиопатия» и «диабетическая кардиомиопатия» в литературе чаще используются как патофизиологические, а не клинические понятия; они признаются полезными для более четкого понимания механизмов развития СН при СД2 [12]. Вполне понятно, что на практике их разграничение затруднительно, так как у каждого конкретного больного с диабетом и СН они, более вероятно, сосуществуют вместе, при этом относительная весомость каждой из них широко варьирует.
Этиология диабетической кардиомиопатии многофакторная. Современные гипотезы развития включают в себя инсулинорезистентность (ИР), эндотелиальную дисфункцию, фиброз, липотоксичность и энергетические нарушения [13].
Согласно литературным данным, ИР играет существенную роль в развитии атеросклероза и артериальной гипертензии [14]. ИР классически определяется как неспособность инсулина стимулировать его метаболическое действие в органах, таких как жировая ткань, скелетные мышцы и печень. Резистентность к инсулину является ранним признаком ожирения, СД2 и, возможно, фактором риска развития СН. Однако вопрос о том, могут ли ожирение и ИР провоцировать СН, остается спорным. Умеренная ИР оказывает благоприятный защитный эффект на сердце от повреждений, вызванных чрезмерным накоплением метаболитов [15], приводящим к увеличению метаболического стресса и развитию дисфункции миокарда [16].
Основные метаболические эффекты инсулина реализуются через передачу сигналов при связывании лиганда с рецептором инсулина, членом которого является семейство рецепторов тирозинкиназы. После связывания лиганда аутофосфорилированные остатки увеличивают активность тирозинкиназы для белков семейства субстратов инсулина (IRS). В дальнейшем рецептор инсулина взаимодействует с белками IRS, чтобы активировать сеть внутриклеточных сигнальных путей, включая фосфатидилинозитол-3-киназу (PI3K) и протеинкиназу В (Akt) [17]. IRS1 и IRS2 являются двумя наиболее распространенными изоформами в сердце и необходимы для инсулин-опосредованной активации PI3K. PI3K состоит из каталитической субъединицы p110 и регуляторной субъединицы p85, которые катализируют генерацию фосфатидилинозитол (3,4,5)-трифосфата (PIP3) и приводят к активации протеинкиназы B [18]. В дополнение к активации сигнального каскада PI3K/Akt инсулин стимулирует другую систему — митоген-активируемую протеинкиназу (MAPK). В результате чего происходят рост и пролиферация эндотелиальных клеток, усиливается миграция их в интиму, увеличиваются пролиферация гладкомышечных клеток и продукция коллагена, что способствует ускоренному развитию атеросклероза [19].
Таким образом, в состояниях, устойчивых к инсулину, наблюдаются ухудшение инсулин-опосредованной активации глюкозы в мышцах и адипоцитах, подавление печеночной продукции и нарушение липолиза [20].
Основными типами клеток в сосудистом русле, в которых была оценена передача сигналов инсулина, являются эндотелиальные и гладкомышечные клетки. В эндотелиальных клетках передача сигналов инсулина через PI3K и Akt приводит к активации эндотелиальной NO-синтазы (eNOS), которая способствует не только снижению тонуса сосудов, но может также активировать противовоспалительные и антиатеросклеротические пути. Таким образом, нарушенная передача сигналов инсулина способствует развитию ожирения и связанной с диабетом эндотелиальной дисфункции, а также гипертонии и ускоренному атеросклерозу [21] .
Важно отметить, что при ИР нарушение активности eNOS может происходить независимо от дефектов в передаче инсулина [22, 23]. Одним из важных моментов в понимании ослабленной передачи сигналов эндотелиального инсулина в контексте ИР является концепция несбалансированной передачи сигналов инсулина, в результате чего периферическая гиперинсулинемия приводит к активации сигнального пути, в то время как другие остаются подавленными. Например, передача сигналов инсулина PI3K-Akt и eNOS может быть нарушена в эндотелиальных клетках, при этом активация сигнальных путей MAPK приводит к экспрессии эндотелина-1 и долгое время остается активной, что способствует увеличению вазоконстрикции [24][25].
Предполагается, что гиперинсулинемия, сопровождающая ИР, вносит существенный вклад в гиперплазию гладкомышечных клеток, способствуя травмированию интимы и тем самым развитию стенозов в артериях [26]. Роль нарушения передачи сигналов инсулина в коронарном русле и патофизиология СН при инсулинорезистентных состояниях изучены недостаточно. Однако для пациентов с ИР и диабетом было показано, что эндотелий-зависимая вазодилатация значительно снижена [27]. Таким образом, существует вероятность того, что нарушение коронарной вазодилатации при инсулинорезистентных состояниях способствует ухудшению сократительного резерва миокарда при СН, особенно у пациентов с ИР.
Эндотелиальная дисфункция является самым ранним признаком сосудистых изменений и представляет собой состояние, приводящее к выработке медиатора оксида азота (NO), образующегося из аминокислоты L-аргинина с участием eNOS. При отсутствии адекватных уровней L-аргинина eNOS генерирует активные формы кислорода (АФК) вместо азотных частиц.
При СД2 ИР и компенсаторная гиперинсулинемия вызывают дисбаланс в активности eNOS путем уменьшения количества кофакторов eNOS. Это приводит к уменьшению вазорелаксации, повышенной экспрессии молекул адгезии, таких как молекула адгезии сосудистого эндотелия 1 типа (VCAM-1) и E-селектин, которые обладают более выраженными проатерогенными и воспалительными свойствами. Позднее при прогрессировании заболевания высокая гипергликемия способствует неферментативному образованию конечных продуктов гликирования и активирует матриксные металлопротеиназы, участвующие в разрыве бляшек и ремоделировании артерий [28]. Длительное воздействие гипергликемии может изменить экспрессию генов в клетках эндотелия, гладких мышц, сетчатки глаза и сердца без изменений в последовательности ДНК.
Хроническая гипергликемия вызывает гликирование митохондриальных белков, что способствует снижению митохондриальной функции и чрезмерной продукции АФК [29]. Митохондриальные белки дыхательной цепи, которые подвергаются гликированию, становятся более склонными к образованию супероксидного аниона, независимо от уровня гипергликемии, повреждающей митохондриальную ДНК, считающуюся даже более чувствительной к окислительным повреждениям, чем ядерная ДНК [30]. Долгосрочное сохранение этих эпигенетических аномалий, которые могут стать необратимыми, представляет собой ключевой механизм, лежащий в основе феномена «метаболической памяти», который относится к измененной экспрессии генов, ответственной за прогрессирование наиболее опасных микро- и макрососудистых диабетических осложнений [31][32].
Понятие «метаболическая память» возникает из экспериментальных и клинических наблюдений, в которых установлено, что раннее воздействие гипергликемической среды «регистрируется» клетками и может объяснить сосудистые осложнения, наблюдаемые у пациентов с диабетом, у которых гликемический контроль не осуществлялся [32]. Поскольку молекулярные изменения касаются в основном эндотелия, наиболее подходящим выражением этого феномена можно считать «эндотелиальная гипергликемическая память».
Повреждение, вызванное гипергликемией, может быть ограничено или предотвращено, когда гликемический контроль достигается на ранней стадии, но его трудно изменить. Если неудовлетворительный контроль длится дольше, это означает, что высокий уровень глюкозы может вызвать различные изменения, сохраняющиеся в течение нескольких дней после нормализации уровня глюкозы.
Гипергликемия способствует развитию сердечно-сосудистых событий, но ее возможно контролировать фармакологически с помощью стандартной терапии гипогликемическими средствами, а также модификацией диеты и физическими упражнениями, но все же у некоторых пациентов продолжают развиваться опасные для жизни сосудистые осложнения, для которых современные методы лечения не всегда эффективны [33].
В возникновении индивидуальных различий в ответной реакции организма на повреждающее действие гипергликемии рассматриваются потенциальная роль генетических полиморфизмов и их влияние на всасывание, биодоступность, эффективность и безопасность антидиабетических препаратов. Наличие большинства однонуклеотидных полиморфизмов (SNP) в некодирующих областях генома или в других регуляторных областях связано с заболеваемостью диабетом [34].
В связи с чем ожидается, что оценка ассоциации эпигенотипов с помощью исследований по всему эпигеному может представлять новую важную информацию о патогенезе диабетических осложнений и метаболической памяти, а также способна определить новые терапевтические методы и диагностические биомаркеры для более раннего вмешательства у этой тяжелой категории пациентов.
Эпигенетические исследования эндотелиальной дисфункции, связанной с диабетом, несомненно, могут быть многообещающей стратегией для выявления пациентов с большей восприимчивостью к развитию микро- и макрососудистых осложнений. Одновременно включение эпибиомаркеров может заложить основу для нового терапевтического подхода, который выбирается индивидуально для каждого больного диабетом в качестве более подходящей терапии с целью увеличения выживаемости и продолжительности жизни.
Клинически диабетическое сердце характеризуется диастолической дисфункцией с сохраненной фракцией выброса ЛЖ. Эти изменения вызваны патологическим ремоделированием сердца вследствие увеличения интерстициального и периваскулярного фиброза и развитием гипертрофии ЛЖ. Увеличение фиброза является результатом повышенного отложения коллагена в сочетании с отклонениями в структуре внеклеточного матрикса [35, 36].
Метаболическая дисфункция, гиперинсулинемия, окислительный стресс и воспаление являются одними из распространенных причин гипертрофии ЛЖ, наблюдаемой у больных диабетом [37]. Гипергликемия, гиперинсулинемия и ИР приводят к увеличению количества окисленных свободных жирных кислот (СЖК), провоспалительных цитокинов, а также накоплению конечных продуктов гликирования. Эти нарушения способствуют изменению метаболизма, приводящему к апоптозу кардиомиоцитов и фиброзу (рис. 1).
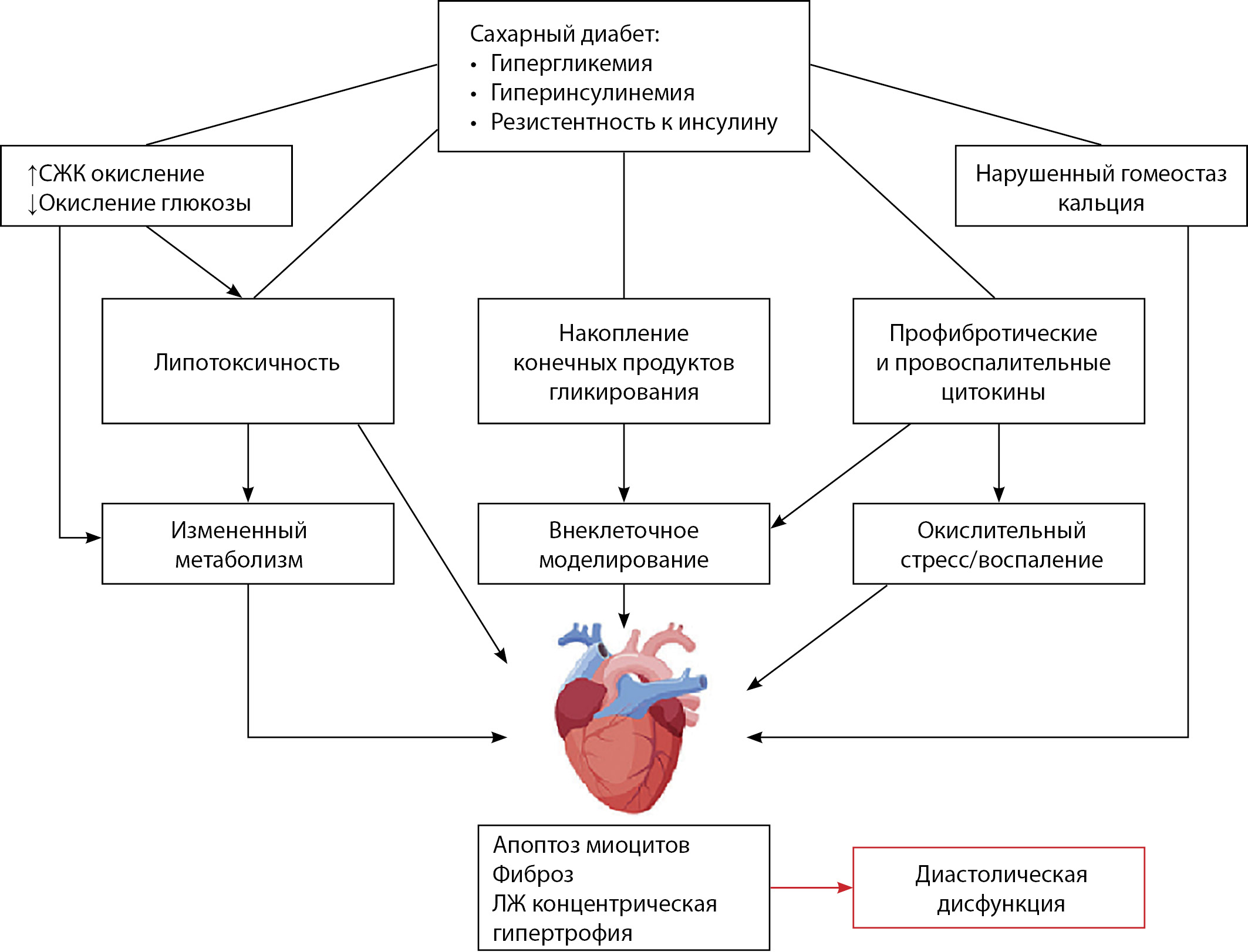
Рисунок 1. Патофизиологические механизмы развития диабетической кардиомиопатии.
Примечание. СЖК — свободные жирные кислоты, ЛЖ — левый желудочек
Прогрессирование диабетической кардиомиопатии, которая, особенно на ранних стадиях, часто носит бессимптомный характер, происходит в несколько разных фаз. Процесс начинается на субклеточном уровне и проявляется ремоделированием миокарда с развитием гипертрофии ЛЖ.
Следующие патофизиологические изменения — это развитие интерстициального и периваскулярного фиброза, которые являются более поздним этапом в прогрессировании заболевания. Гипертрофия и фиброз вызывают нарушения расслабления, пассивное наполнение ЛЖ и, как следствие, диастолическую дисфункцию ЛЖ.
Как упоминалось ранее, диастолическая дисфункция представляет собой основные функциональные нарушения у больных диабетом, которые могут протекать бессимптомно на ранних стадиях [38].
Систолическая дисфункция развивается реже и только у небольшого процента пациентов на поздних стадиях диабетической кардиомиопатии [39]. Кроме того, связанный с диабетом фиброз способствует развитию фибрилляции предсердий и внезапной смерти [40].
В настоящее время нет конкретных морфологических изменений, биохимических маркеров или клинических проявлений, необходимых для постановки диагноза диабетической кардиомиопатии. Эта патология на ранних стадиях часто имеет бессимптомный характер и обычно сопровождается другими осложнениями, что делает окончательный диагноз сложным.
В условиях ИР адипоциты не способны реагировать на стимулы инсулина и подавлять липолиз. Одним из последствий ИР является высвобождение жировыми клетками большого количества СЖК в кровоток. Таким образом, инициируются системные липотоксические эффекты, включая эктопическое отложение липидов в тканях организма, а также образование множества токсических медиаторов (ацил-КоА, церамид и диацилглицерин) и последующее прерывание передачи сигналов инсулина. Это приводит к липотоксичности и в дальнейшем усугублению ИР [41].
Важно подчеркнуть, что избыток липидов вреден в случае наличия насыщенных СЖК, тогда как моно- и полиненасыщенные СЖК часто оказывают антилипотоксическое действие. Наиболее опасна насыщенная СЖК, найденная в плазме, такая как пальмитиновая кислота, которая вызывает окислительный стресс через β-окисление в митохондриях и приводит к стрессу в эндоплазматическом ретикулуме и нарушению в Ca2+ гомеостазе. Таким образом, липотоксичность, вызванная насыщенными СЖК, способствует дисфункции митохондрий, усугубляет окислительный стресс и ИР, вызывая тем самым воспаление [42][43].
Эти процессы, посредством которых СЖК вызывает токсичность, имеют место в нескольких тканях организма, в том числе и в β-клетках поджелудочной железы. Было показано, что высокие концентрации СЖК в β-клетках способствуют пролиферации β-клеток, при этом хроническое липотоксическое состояние, возникающее при диабете, может способствовать нарушению секреции инсулина и в конечном итоге вызывать гибель β-клеток [44]. Наконец, окислительный стресс, вызванный митохондриями и дисфункцией эндоплазматического ретикулума, также связан с липотоксичностью в β-клетках (рис. 2).
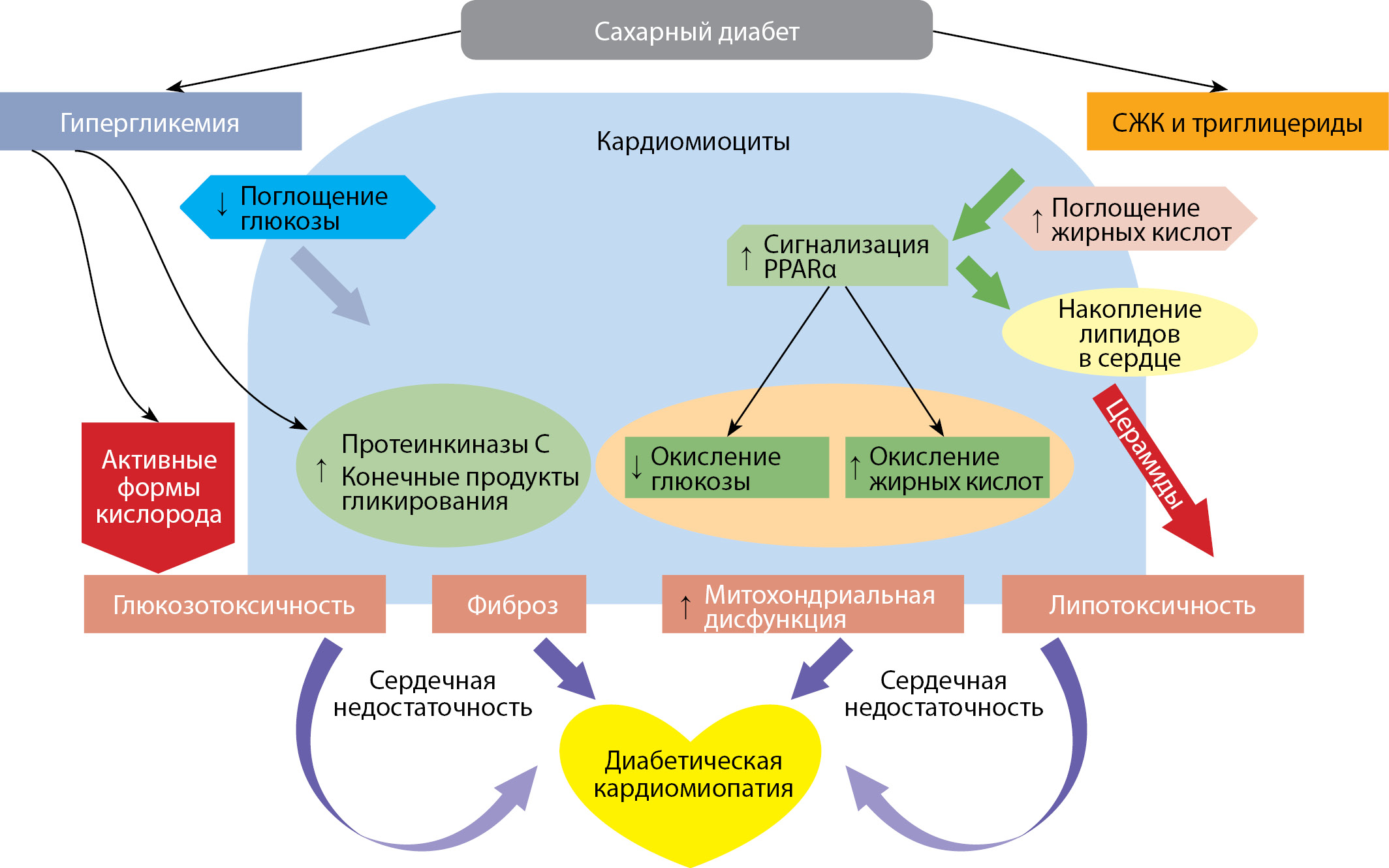
Рисунок 2. Процессы, приводящие к развитию диабетической кардиомиопатии.
Таким образом, митохондриальная дисфункция и существование окислительного стресса, по-видимому, обусловливают роль липотоксичности в развитии диабета, благодаря которой он усугубляет ИР и недостаточность β-клеток поджелудочной железы.
Сердце преобразует химическую энергию, присутствующую в форме субстратов и кислорода, в механическую энергию и тепло [45]. Поддержание адекватного уровня сердечной деятельности происходит с помощью фосфатных метаболитов, аденозинтрифосфата (АТФ), фосфокреатина, которые являются основными компонентами для накопления энергии [46]. Для получения АТФ здоровое сердце способно метаболизировать ряд субстратов, включая СЖК, глюкозу, аминокислоты, кетоны и лактат. На нормальное сердце приходится всего около 0,5% общей массы человеческого тела, при этом доля АТФ, потребляемого ежедневно, достигает 8% [47]. Кроме того, потребление энергии увеличивается в геометрической прогрессии под воздействием сердечного стресса [48]. Следовательно, недостаточное энергоснабжение миокарда, нарушение утилизации субстрата и окислительный стресс считаются ответственными за прогрессирование СН [49]. Однако представляется затруднительным рассматривать СН с метаболической точки зрения, учитывая гибкость метаболизма сердечного субстрата (способность организма реагировать на изменения метаболических или энергетических потребностей) и сложной метаболической сети [50].
Согласно литературным данным, митохондриальная дисфункция вносит свой вклад в этиологию СН [51]. Следует отметить, что умеренная митохондриальная дисфункция или стресс снижают факторы риска развития СН частично за счет метаболической регуляции митокинов. Митокины — это тип цитокинов, пептидов или сигнальных путей, продуцируемых или активируемых ядром или митохондриями через клеточные неавтономные реакции во время клеточного стресса. В дополнение к стимулированию связи между митохондриями и ядром митокины также оказывают системное регуляторное действие, циркулируя в отдаленных тканях. В настоящее время появляется все больше данных, свидетельствующих о том, что митокины способны снижать метаболические факторы риска и связаны со степенью тяжести СН [52]. Следовательно, митокины могут представлять собой потенциальную мишень для терапии СН. Кроме того, митокины способны улучшать клеточный метаболизм с помощью непрямых факторов, таких как ингибирование воспаления, ослабление повреждающего действия окислительного стресса, уменьшение аутофагии и задержки клеточного старения [53–55].
В дополнение к этим системным нарушениям, ИР связана с изменениями в метаболической среде, такими как низкоинтенсивное воспаление с увеличением липопротеинассоциированной фосфолипазы А2, повышение концентраций различных цитокинов и измененная секреция адипокинов, в том числе лептина, резистина и адипонектина [56–58].
Воспаление может сопутствовать ИР, сопровождаясь повышением уровня циркулирующих провоспалительных цитокинов — фактора некроза опухоли (ФНО) альфа, интерлейкинов (ИЛ) ИЛ-1 и -6, а также снижением содержания противовоспалительных медиаторов, таких как ИЛ-10 и ФНО1β [59]. Согласно литературным данным, уровень воспалительных цитокинов повышен у пациентов с СН [60][61].
Данные факторы (ФНОα, ИЛ-6 и СЖК) механически активируют внутриклеточные киназы, которые потенцируют фосфорилирование белков IRS, тем самым ослабляя передачу сигналов инсулина и индуцируя сопротивление инсулина. Такая сложная патофизиология ИР тесно взаимосвязана с ожирением и малоподвижным образом жизни. Большинство исследований, касающихся механизмов ИР, было выполнено у пациентов с ожирением или СД, при этом меньшее количество исследований было проведено у пациентов с СН. ИР при СН также коррелирует с повышением сывороточной концентрации провоспалительных цитокинов, катехоламинов, катаболических стероидов и гормона роста [62].
Несмотря на растущий интерес к изучению диабетической кардиомиопатии, на сегодняшний день отсутствует четкое определение данного патологического состояния. Вместе с тем в настоящее время доказана целесообразность применения как при диабетической кардиомиопатии, так и при СН у больных с СД, имеющих общие механизмы формирования, стратегии многофакторного воздействия, при которой, наряду с адекватным контролем углеводного обмена, основополагающей целью является снижение сердечно-сосудистого риска и смертности.
Проблема сочетания СН и СД2 многогранна и требует дальнейшего продолжения исследований. Среди нерешенных проблем остаются такие вопросы, как возможность обратимости изменений при диабетической кардиомиопатии; каковы оптимальные целевые уровни гликированного гемоглобина для больных с разными стадиями СН; безопасность сахароснижающих препаратов у лиц с диабетом и хронической СН с сохраненной и низкой фракцией выброса ЛЖ.
Последнее стало возможным с внедрением в клиническую практику нового класса сахароснижающих препаратов — ингибиторов натрий-глюкозного котранспортера 2-го типа, подтвердивших высокую эффективность в способности снижения риска сердечно-сосудистой смертности и госпитализаций по поводу СН у больных СД и ассоциированными ССЗ, что, безусловно, будет способствовать улучшению прогноза.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Финансирование. Тема фундаментальных научных исследований «Изучение механизмов структурного и функционального ремоделирования миокарда при разных фенотипах хронической сердечной недостаточности ишемической и неишемической этиологии» № 122020300045-5.
Участие авторов. Сваровская А.В. — разработка концепции и дизайна; Гарганеева А.А. — проверка критически важного интеллектуального содержания, окончательное утверждение для публикации рукописи. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
1. Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of Amer. Circulation. 2017;136(6):86-92. doi: https://doi.org/10.1161/CIR.0000000000000509
2. Metra M, Teerlink JR. Heart failure. Lancet. 2017;390:1981-1995. doi: https://doi.org/10.1016/S0140-6736(17)31071-1.
3. Lara KM, Levitan EB, Gutierrez OM, et al. Dietary Patterns and Incident Heart Failure in U.S. Adults Without Known Coronary Disease. J Am Coll Cardiol. 2019;73:2036-2045. doi: https://doi.org/10.1016/j.jacc.2019.01.067
4. Brown DA, Perry JB, Allen ME, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14:238-250. doi: https://doi.org/10.1038/nrcardio.2016.203
5. Cavender MA, Steg PG, Smith SC Jr, et al. Impact of Diabetes Mellitus on Hospitalization for Heart Failure, Cardiovascular Events, and Death: Outcomes at 4 Years From the Reduction of Atherothrombosis for Continued Health (REACH) Registry. Circulation. 2015;132(10):923-931. doi: https://doi.org/10.1161/CIRCULATIONAHA.114.014796
6. Paneni F. Empaglifl ozin across the stages of diabetic heart disease. Eur Heart J. 2018;39(5):371-373. doi: https://doi.org/10.1093/eurheartj/ehx519
7. Lee WS, Kim J. Diabetic cardiomyopathy: where we are and where we are going. Korean J Intern Med. 2017;32:404-421. doi: https://doi.org/10.3904/kjim.2016.208
8. Резник Е.В., Никитин И.Г. Кардиоренальный синдром у больных с сердечной недостаточностью как этап кардиоренального континуума (часть I): определение, классификация, патогенез, диагностика, эпидемиология (обзор литературы) // Архив Внутренней Медицины. — 2019. — Т. 9. — №1. — С. 5-22. doi: https://doi.org/10.20514/2226-6704-2019-9-1-5-22
9. Котов С.В., Рудакова И.Г., Исакова Е.В., Волченкова Т.В. Диабетическая нейропатия: разнообразие клинических форм (лекция) // РМЖ. Медицинское обозрение. — 2017. — Т. 25. — №11. — С. 822-830.
10. Spallone V, Ziegler D, Freeman R, et al. Cardiovascular autonomic neuropathy in diabetes: clinical impact, assessment, diagnosis, and management. Diabetes Metab Res Rev. 2011;27(7):639-653. doi: https://doi.org/10.1002/dmrr.1239
11. Багрий А.Э., Супрун Е.В., Михайличенко Е.С., Голодников И.А. Хроническая сердечная недостаточность и сахарный диабет 2 типа: состояние проблемы // Российский кардиологический журнал. — 2020. — Т. 25. — №4. — С. 3858. doi: https://doi.org/10.15829/1560-4071-2020-3858
12. Lorenzo-Almorós A, Tuñón J, Orejas M, et al. Diagnostic approaches for diabetic cardiomyopathy. Cardiovascular diabetology. 2017;16(28):1-14. doi: https://doi.org/10.1186/s12933-017-506-x
13. Althunibat OY, Al Hroob AM, Abukhalil MH, et al. Fisetin ameliorates oxidative stress, inflammation and apoptosis in diabetic cardiomyopathy. Life Sci. 2019;221:83-92. doi: https://doi.org/10.1016/j.lfs.2019.02.017.
14. Di Pino A, DeFronzo RA. Insulin Resistance and Atherosclerosis: Implications for Insulin Sensitizing Agents. Endocr Rev. 2019;40(6):1447-1467. doi: https://doi.org/10.1210/er.2018-00141
15. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. 2018;15:457-470. doi: https://doi.org/10.1038/s41569-018-0044-6
16. Ceylan-Isik AF, Kandadi MR, Xu X, et al. Apelin administration ameliorates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Cardiol. 2013;63:4-13. doi: https://doi.org/10.1016/j.yjmcc.2013.07.002
17. Kadowaki T, Ueki K, Yamauchi T, Kubota N. SnapShot: Insulin Signaling Pathways. Cell. 2012;148(3):624-624.e1. doi: https://doi.org/10.1016/j.cell.2012.01.034
18. Демидова Т.Ю., Зенина С.Г. Роль инсулинорезистентности в развитии сахарного диабета и других состояний. Современные возможности коррекции // РМЖ. Медицинское обозрение. — 2019. — Т. 10. — №II. — С. 116-122.
19. Riehle C, Abel ED. Insulin Signaling and Heart Failure. Circ Res. 2016;118(7):1151-1169. doi: https://doi.org/10.1161/CIRCRESAHA.116.306206
20. Boucher J, Kleinridders A, Kahn CR. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb Perspect Biol. 2014;6(1):a009191-a009191. doi: https://doi.org/10.1101/cshperspect.a009191
21. Symons JD, Abel ED. Lipotoxicity contributes to endothelial dysfunction: A focus on the contribution from ceramide. Rev Endocr Metab Disord. 2013;14(1):59-68. doi: https://doi.org/10.1007/s11154-012-9235-3
22. Bharath LP, Ruan T, Li Y, et al. Ceramide-Initiated Protein Phosphatase 2A Activation Contributes to Arterial Dysfunction In Vivo. Diabetes. 2015;64(11):3914-3926. doi: https://doi.org/10.2337/db15-0244
23. Zhang QJ, Holland WL, Wilson L, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by pp2a-mediated dephosphorylation of the enos-akt complex. Diabetes. 2012;61:1848-1859. doi: https://doi.org/10.2337/db11-1399.
24. Li Q, Park K, Li C, et al. Induction of vascular insulin resistance and endothelin-1 expression and acceleration of atherosclerosis by the overexpression of protein kinase c-beta isoform in the endothelium. Circulation research. 2013;113(4):418-427. doi: https://doi.org/10.1161/CIRCRESAHA.113.301074
25. Park K, Li Q, Rask-Madsen C, et al. Serine phosphorylation sites on irs2 activated by angiotensin ii and protein kinase c to induce selective insulin resistance in endothelial cells. Molecular and cellular biology. 2013;33(16):3227-3241. doi: https://doi.org/10.1128/MCB.00506-13.
26. Isenovic E, Kedees M, Tepavcevic S, et al. Role of PI3K/AKT, cPLA2 and ERK1/2 Signaling Pathways in Insulin Regulation of Vascular Smooth Muscle Cells Proliferation. Cardiovasc Hematol Disord Targets. 2009;9(3):172-180. doi: https://doi.org/10.2174/187152909789007034
27. Стаценко М.Е., Деревянченко М.В. Роль системного воспаления в снижении эластичности магистральных артерий и прогрессировании эндотелиальной дисфункции у больных артериальной гипертензией в сочетании с ожирением, сахарным диабетом 2 типа // Российский кардиологический журнал. — 2018. — Т. 23. — №4. — С. 32–36. doi: https://doi.org/10.15829/1560-4071-2018-4-32-36.
28. Fishman SL, Sonmez H, Basman C, et al. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol. Med. 2018;24:59. doi: https://doi.org/10.1186/s10020-018-0060-3
29. Rajasekar P, O’Neill CL, Eeles L, et al. Epigenetic Changes in Endothelial Progenitors as a Possible Cellular Basis for Glycemic Memory in Diabetic Vascular Complications. J Diabetes Res. 2015;2015(3):1-17. doi: https://doi.org/10.1155/2015/436879
30. Berezin A. Metabolic memory phenomenon in diabetes mellitus: Achieving and perspectives. Diabetes Metab Syndr Clin Res Rev. 2016;10(2):S176-S183. doi: https://doi.org/10.1016/j.dsx.2016.03.016
31. Prattichizzo F, Giuliani A, Ceka A, et al. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin Epigenetics. 2015;7(1):56. doi: https://doi.org/10.1186/s13148-015-0090-4
32. Potenza MA, Nacci C, De Salvia MA, et al. Targeting endothelial metaflammation to counteract diabesity cardiovascular risk: Current and perspective therapeutic options. Pharmacol Res. 2017;120(1):226-241. doi: https://doi.org/10.1016/j.phrs.2017.04.009
33. Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190-1195. doi: https://doi.org/10.1126/science.1222794.
34. Reddy MA, Natarajan R. Role of Epigenetic Mechanisms in the Vascular Complications of Diabetes. Pharmacological Research. 2013;120:435-454. doi: https://doi.org/10.1007/978-94-007-4525-4_19
35. Tate M, Grieve DJ, Ritchie RH. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin Sci. 2017;131(10):897-915. doi: https://doi.org/10.1042/CS20160491
36. Velic A, Laturnus D, Chhoun J, et al. Diabetic Basement Membrane Thickening Does Not Occur in Myocardial Capillaries of Transgenic Mice When Metallothionein is Overexpressed in Cardiac Myocytes. Anat Rec. 2013;296(3):480-487. doi: https://doi.org/10.1002/ar.22646
37. Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: Mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. 2014;142(3):375-415. doi: https://doi.org/10.1016/j.pharmthera.2014.01.003
38. Borghetti G, von Lewinski D, Eaton DM, et al. Diabetic Cardiomyopathy: Current and Future Therapies. Beyond Glycemic Control. Front Physiol. 2018;9(3):375-415. doi: https://doi.org/10.3389/fphys.2018.01514
39. Palomer X, Pizarro-Delgado J, Vázquez-Carrera M. Emerging Actors in Diabetic Cardiomyopathy: Heartbreaker Biomarkers or Therapeutic Targets? Trends Pharmacol Sci. 2018;39(5):452-467. doi: https://doi.org/10.1016/j.tips.2018.02.010
40. Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016;90:84-93. doi: https://doi.org/10.1016/j.yjmcc.2015.12.011
41. Burgos-Morón, Abad-Jiménez, Marañón AM, et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J Clin Med. 2019;8(9):1385. doi: https://doi.org/10.3390/jcm8091385
42. Tumova J, Andel M, Trnka J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol. Res. 2016;65(2):193-207. doi: https://doi.org/10.33549/physiolres.932993
43. Palomer X, Pizarro-Delgado J, Barroso E, Vázquez-Carrera M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol Metab. 2018;29(3):178-190. doi: https://doi.org/10.1016/j.tem.2017.11.009
44. Oh YS, Bae GD, Baek DJ, et al. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front Endocrinol (Lausanne). 2018;9(3):178-190. doi: https://doi.org/10.3389/fendo.2018.00384
45. Taegtmeyer H, Young ME, Lopaschuk GD, et al. Assessing Cardiac Metabolism. Circ Res. 2016;118(10):1659-1701. doi: https://doi.org/10.1161/RES.0000000000000097
46. Peterzan MA, Lygate CA, Neubauer S, Rider OJ. Metabolic remodeling in hypertrophied and failing myocardium: a review. Am J Physiol Circ Physiol. 2017;313(3):H597-H616. doi: https://doi.org/10.1152/ajpheart.00731.2016
47. Brown DA, Perry JB, Allen ME, et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14(4):238-250. doi: https://doi.org/10.1038/nrcardio.2016.203
48. Smyrnias I, Gray SP, Okonko DO, et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J Am Coll Cardiol. 2019;73(14):1795-1806. doi: https://doi.org/10.1016/j.jacc.2018.12.087
49. Münzel T, Camici GG, Maack C, et al. Impact of Oxidative Stress on the Heart and Vasculature. J Am Coll Cardiol. 2017;70(2):212-229. doi: https://doi.org/10.1016/j.jacc.2017.05.035
50. Bertero E, Maack C. Metabolic remodeling in heart failure. Nat Rev Cardiol. 2018;15:457-470. doi: https://doi.org/10.1038/s41569-018-0044-6
51. Sorrentino V, Menzies KJ, Auwerx J. Repairing Mitochondrial Dysfunction in Disease. Annu Rev Pharmacol Toxicol. 2018;58:353–89. doi: https://doi.org/10.1146/annurev-pharmtox-010716-104908
52. Kim KH, Jeong YT, Oh H, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83-92. doi: https://doi.org/10.1038/nm.3014
53. Foote K, Bennett MR. Molecular insights into vascular aging. Aging (Albany NY). 2018;10:3647-3649. doi: https://doi.org/10.18632/aging.101697
54. Crupi AN, Nunnelee JS, Taylor DJ, et al. Oxidative muscles have better mitochondrial homeostasis than glycolytic muscles throughout life and maintain mitochondrial function during aging. Aging (Albany NY). 2018;10:3327-3352. doi: https://doi.org/10.18632/aging.101643
55. Fuku N, Pareja-Galeano H, Zempo H, et al. The mitochondrial-derived peptide MOTS-c: a player in exceptional longevity? Aging Cell. 2015;14:921-923. doi: https://doi.org/10.1111/acel.12389
56. Keane KN, Cruzat VF, Carlessi R, et al. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and β -Cell Dysfunction. Oxid Med Cell Longev. 2015;2015(2):1-15. doi: https://doi.org/10.1155/2015/181643
57. Тепляков А.Т., Кузнецова А.В., Протопопова Н.В., и др. Прогностическое значение липопротеин-ассоциированной фосфолипазы А2 в стратификации сердечно-сосудистого риска после коронарного стентирования у пациентов с СД 2 типа: какой порог решающего правила выбрать? // Бюллетень сибирской медицины. — 2015. — Т. 14. — №2. — С. 47-54. doi: https://doi.org/10.20538/1682-0363-2015-2-47-54
58. Blüher M. Adipokines – removing road blocks to obesity and diabetes therapy. Mol Metab. 2014;3(3):230-240. doi: https://doi.org/10.1016/j.molmet.2014.01.005
59. Сваровская А.В., Тепляков А.Т. Инсулинорезистентность при сахарном диабете. Контроль над риском кардиоваскулярных осложнений. — Томск: НИИ кардиологии, Томский НИМЦ; 2018. — 196 с.
60. Тепляков А.Т., Болотская Л.А., Дибиров М.М., и др. Клиникоиммунологические нарушения у больных с постинфарктным ремоделированием левого желудочка с хронической сердечной недостаточностью // Терапевтический архив. — 2008. — Т. 80. — №11. — С. 52-57.
61. Fiordelisi A, Iaccarino G, Morisco C, et al. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int J Mol Sci. 2019;20(7):1599. doi: https://doi.org/10.3390/ijms20071599
62. Токмачев Р.Е., Будневский А.В., Кравченко А.Я. Роль воспаления в патогенезе хронической сердечной недостаточности // Терапевтический архив. — 2016. — Т. 88. — №9. — С. 106-110. doi: https://doi.org/10.17116/terarkh2016889106-110
Сваровская Алла Владимировна, доктор медицинских наук , старший научный сотрудник
634012, Томск, Киевская ул., д. 111А
eLibrary SPIN: 2390-2877
конфликт интересов отсутствует
Гарганеева Алла Анатольевна, доктор медицинских наук, профессор
Томск
eLibrary SPIN: 6774-7931
конфликт интересов отсутствует
|
|
1. Рисунок 1. Патофизиологические механизмы развития диабетической кардиомиопатии. Примечание. СЖК — свободные жирные кислоты, ЛЖ — левый желудочек | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(319KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Процессы, приводящие к развитию диабетической кардиомиопатии. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(367KB)
|
Метаданные ▾ | |
Сваровская А.В., Гарганеева А.А. Сахарный диабет 2 типа и сердечная недостаточность — современный взгляд на механизмы развития. Сахарный диабет. 2022;25(3):267-274. https://doi.org/10.14341/DM12648
Svarovskaya A.V., Garganeeva A.A. Diabetes mellitus and heart failure — a modern look at the mechanisms of development. Diabetes mellitus. 2022;25(3):267-274. (In Russ.) https://doi.org/10.14341/DM12648

 |
Адрес: 117036, Российская Федерация, Москва, улица Дмитрия Ульянова, дом 11.
Обработка персональных данных







































