




Содержание
Перейти к:
https://doi.org/10.14341/DM13102
Перейти к:
Синдромы липодистрофии — это гетерогенная группа крайне редких, наследственных или приобретенных заболеваний, которые характеризуются полной или частичной потерей подкожной жировой клетчатки (ПЖК) или неправильным ее перераспределением. Распространенность липодистрофий оценивается как 1:1 000 000 населения, в настоящее время в литературе описано около 1000 случаев.
Саркоидоз — мультисистемное заболевание, характеризующееся образованием неказеифицирующих эпителиоидно-клеточных гранулем в пораженных тканях. Несмотря на большое количество проведенных исследований, этиология и патогенез саркоидоза все еще остаются неизвестными. Большинство исследователей ссылаются на возможный аутоиммунный или иммуноопосредованный генез заболевания.
В данной статье представлены уникальные клинические случаи сочетания двух редких заболеваний у одного пациента: саркоидоза и семейной парциальной липодистрофии.
Фролкова Н.В., Кокшарова Е.О., Васильев П.А., Смирнова О.М., Шестакова М.В. Сочетание семейной парциальной липодистрофии (синдром Даннигана - Кобберлинга) с саркоидозом легких. Сахарный диабет. 2024;27(3):287-294. https://doi.org/10.14341/DM13102
Frolkova N.V., Koksharova E.O., Vasiluev P.A., Smirnova O.M., Shestakova M.V. Combination of familial partial lipodystrophy (Dunnigan-Cobberling syndrome) with pulmonary sarcoidosis. Diabetes mellitus. 2024;27(3):287-294. (In Russ.) https://doi.org/10.14341/DM13102
Синдромы липодистрофии — это гетерогенная группа крайне редких, наследственных или приобретенных заболеваний, которые характеризуются генерализованной (затрагивает почти все жировые депо тела) или парциальной (затрагивает только конечности, или верхнюю часть, или небольшие участки тела) потерей подкожной жировой клетчатки (ПЖК), а также неправильным ее перераспределением, при условии отсутствия предшествующего голодания или катаболического состояния [1–3]. Наследственные липодистрофии ассоциированы с наличием мутации в одном из генов, ответственных за адипогенез, дифференцировку и регуляцию апоптоза адипоцитов. Пациенты имеют предрасположенность к развитию метаболических осложнений, таких как сахарный диабет, гипертриглицеридемия, стеатоз печени, синдром поликистозных яичников и черный акантоз (acanthosis nigricans) [2][3].
Саркоидоз — это мультисистемное воспалительное заболевание неустановленной этиологии, характеризующееся наличием неказеозных эпителиоидных гранулем наряду со скоплением Т-лимфоцитов и макрофагов в пораженных органах [4][6][7]. В дополнение к генетической предрасположенности такие триггеры, как инфекция, неорганические материалы и факторы окружающей среды, вероятно, играют определенную роль, что приводит к возможному развитию аутоиммунного ответа при этом заболевании [5]. Саркоидоз легких может сосуществовать с другими аутоиммунными заболеваниями, что предполагает возможность общего патогенеза и генетической предрасположенности [5].
В данной статье мы представляем уникальные клинические случаи пациентов с сочетанием двух орфанных заболеваний: саркоидоза и семейной парциальной липодистрофии (3 и 4 типов) (перечень редких (орфанных) заболеваний Минздрава России, от 06 февраля 2023 г.).
Ранее приведенный клинический случай был подробно описан Соркиной Е.Л. и соавт. [8].
Пациентка К., 35 лет, с семейной парциальной липодистрофией 3 типа вследствие мутации p.R212Q в гене PPARG поступила в отделение терапии диабета ФГБУ «НМИЦ эндокринологии» МЗ РФ в июне 2022 г. с жалобами на боли в нижних конечностях, снижение остроты зрения, тяжесть в правом подреберье.
Из анамнеза известно, что раннее развитие нормальное, менархе с 13 лет, менструальный цикл нерегулярный.
С 20 лет отметила избыточный рост волос в области верхней губы, подбородка, спины, белой линии живота. В 23 года по поводу олигоменореи принимала комбинированный препарат этинилэстрадиола и дроспиренона в течение 6 месяцев. Впоследствии отметила появление множественных ксантом на ягодицах, спине, плечах, впервые были выявлены дислипидемия и гепатоспленомегалия.
С 25 лет пациентка наблюдалась в клинике эндокринологии Первого МГМУ им. И.М. Сеченова по поводу синдрома поликистозных яичников (СПЯ) с инсулинорезистентностью (ИР), нарушения менструального цикла по типу олигоменореи, отсутствия беременности в течение 2 лет половой жизни без предохранения.
В возрасте 25 лет с жалобами на боли в правом подреберье находилась на обследовании в гастроэнтерологическом отделении, где были впервые диагностированы неалкогольный стеатогепатит, хронический холецистит. Впоследствии при МР-спектроскопии печени (на базе ФГБУ РКНПК МЗ РФ) в двух объемах в 6 и 7 сегментах левой доли процентное содержание жировой ткани составило 38 и 56% соответственно (норма до 6,5%), что позволило верифицировать у пациентки жировой гепатоз. В то же время впервые диагностирована гиперинсулинемия (ИРИ — 20 мкЕд/мл при норме до 17 мкЕд/мл) и ИР (расчетный индекс инсулинорезистентности НОМА-IR 4,4, при норме до 2,7), дислипидемия (триглицериды до 10,35 ммоль/л), гиперурикемия [8]. Пациентке был (офф-лейбл) назначен метформин в суточной дозе 1000 мг с положительным эффектом.
С 26 лет отметила повышение АД максимально до 180/100 мм рт.ст., ЧСС до 100–110 уд/мин. Инициирована антигипертензивная терапия [8].
Впервые госпитализирована в ФГБУ «НМИЦ Эндокринологии» МЗ РФ в 2015 г. (28 лет), проведен пероральный глюкозотолерантный тест, диагностировано нарушение толерантности к глюкозе (НТГ). При гормональном обследовании: лептин — 5,3 нг/мл (1,1–27,6); подтверждена гиперандрогения, исключен гиперкортицизм [8].
Обращал на себя внимание семейный анамнез пациентки.
Впервые при осмотре было выявлено характерное перераспределение ПЖК по типу парциальной липодистрофии (липогипертрофии в области лица, шеи, надключичных областях, избыточное развитие в абдоминальной области (окружность талии — 86 см, бедер — 96 см), липодистрофии в области голеней, бедер, ягодиц, живота (рис. 1)), наличие acanthosis nigricans в подмышечных областях (рис. 2), индекс массы тела (ИМТ)=27,5 кг/м2.
Учитывая данные осмотра и обследования, а также семейный анамнез по СД, у пациентки был заподозрен диагноз семейной парциальной липодистрофии.
В ФГБУ ЭНЦ проведен молекулярно-генетический анализ. В гене PPARG (NM_001374266.1) был выявлен вариант нуклеотидной последовательности c.635G>A в гетерозиготном положении, приводящий к миссенс-замене p.Arg212Gln. Аналогичный вариант был выявлен у отца пациентки и его двоюродной сестры (рис. 3). Изначально этот вариант расценивался как вариант неопределенного клинического значения, однако результаты сегрегационного анализа позволяют перевести его в вероятно-патогенный (критерии PM2, PM5, PP2, PP1, PP4) согласно рекомендациям Американского колледжа медицинской генетики и геномики (ACMG) от 2015 г. и руководству по интерпретации данных массового параллельного секвенирования [9].
На основании полученных результатов впервые установлен диагноз «Семейная парциальная липодистрофия 3 типа». Учитывая отсутствие в 2015 г. препаратов для патогенетического лечения этого заболевания, было рекомендовано продолжить прием фенофибрата, метформина, соблюдение диеты со строгим ограничением жиров животного происхождения, легкоусвояемых углеводов, достаточным содержанием пищевых волокон. Рекомендованы регулярные физические нагрузки, как аэробные, так и анаэробные. На фоне лечения отмечалось снижение, но не нормализация уровней общего холестерина, холестерина липопротеинов низкой плотности и триглицеридов, нормализация показателей азотистого обмена.
С июня 2016 по август 2017 гг. пациентка участвовала в международном рандомизированном клиническом исследовании гиполипидемического препарата Воланесорсен для лечения гипертриглицеридемии. Воланесорсен — антисмысловой олигонуклеотид второго поколения, ингибирует синтез аполипопротеина C-III (ApoCIII) в печени путем связывания с иРНК ApoCIII [10]. ApoCIII играет решающую роль в метаболизме триглицеридов, ингибируя липопротеинлипазу и активность печеночной липазы, а также контролируя биосинтез печеночных липопротеинов [10]. На фоне терапии пациентка отметила улучшение метаболических показателей, восстановление менструального цикла, однако применение исследуемого препарата было прекращено досрочно в связи с развитием нежелательных явлений (местных: выраженные гиперемии и аллергические реакции в местах инъекций и стойкой тромбоцитопении).
После окончания участия в исследовании в августе 2017 г. (30 лет) пациентка отметила ухудшение состояния (боль в горле, острый ринит, одышка), в связи с чем была выполнена флюорография органов грудной клетки, выявлены признаки двусторонней диссеминации в легких. На КТ грудной полости: картина диссеминированного процесса в легких с увеличением всех групп внутригрудных лимфоузлов. Проведена биопсия легочной ткани, подтвердившая диагноз «саркоидоз легких», назначен метилпреднизолон 4 мг. Отметила прибавку массы тела на 10 кг. На фоне приема метформина показатели гликемии натощак и HbA1c сохранялись в пределах нормальных значений. На контрольной МСКТ легких патологических изменений не выявлено. Со слов пациентки, диагноз саркоидоза не снят, дистанционно наблюдается у пульмонолога, принимает метилпреднизолон 2 мг ежедневно.
В декабре 2018 г. (31 год) при госпитализации в ФГБУ «НМИЦ эндокринологии» МЗ РФ от 12.2018 г. на фоне отмены метформина в течение недели повторно проведен пероральный глюкозотолерантный тест, по результатам которого установлен диагноз СД. Скорректирована сахароснижающая терапия: метформин с пролонгированным высвобождением 750 мг утром и вечером.
В 2020 и 2021 гг. — замершие беременности на сроке 2 месяцев.
На момент госпитализации в 2022 г. пациентка получала: метформин с пролонгированным высвобождением — 750 мг утром и вечером, аторвастатин — 10 мг, фенофибрат — 145 мг на ночь, бисопролол — 5 мг утром, метилпреднизолон — 2 мг утром.
При осмотре:
По данным проведенного обследования отмечалась удовлетворительная компенсация углеводного обмена (HbA1c — 5,2%). Данных за наличие микро- и макрососудистых осложнений СД не получено. В биохимическом анализе крови сохранялась гипертриглицеридемия до 5,47 ммоль/л, гиперурикемия до 377,54 мкмоль/л. Повторно даны рекомендации по соблюдению диеты со строгим ограничением жиров животного происхождения, пуринов.
Проведен медицинский консилиум, принято решение: пациентке с диагнозом «семейная парциальная липодистрофия 3 типа» и выраженными метаболическими осложнениями прогрессирующего характера с патогенетической целью необходимо назначение терапии рекомбинантным человеческим метионил-лептином.
Диагноз: «Семейная парциальная липодистрофия 3 типа вследствие мутации p.R212Q в гене PPARG. Липоатрофический сахарный диабет. Дислипидемия. Артериальная гипертензия 3 ст., риск ССО 4. Дисфункция яичников репродуктивного периода. Синдром поликистозных яичников. Гирсутизм. Неалкогольная жировая болезнь печени. Стеатогепатоз. Гиперурикемия. Мочекаменная болезнь: микролиты почек. Саркоидоз легких и внутригрудных лимфатических узлов, морфологически верифицированный, регрессирующее течение».
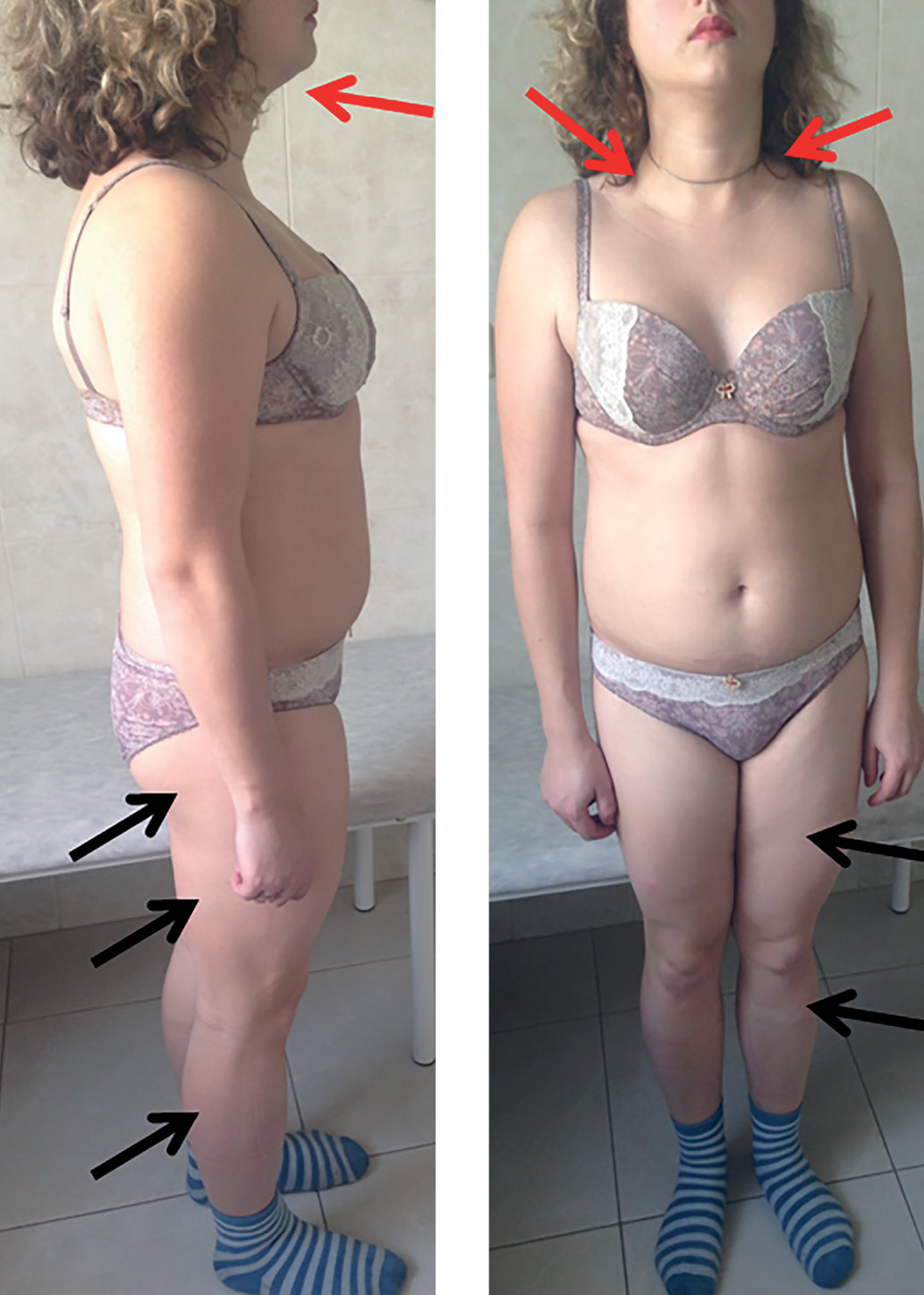
Рисунок 1. Фенотип пациентки К. Красной стрелкой обозначены зоны липогипертрофии; черной — зоны липодистрофии (впервые опубликовано Соркина Е.Л. и соавт.) [8].
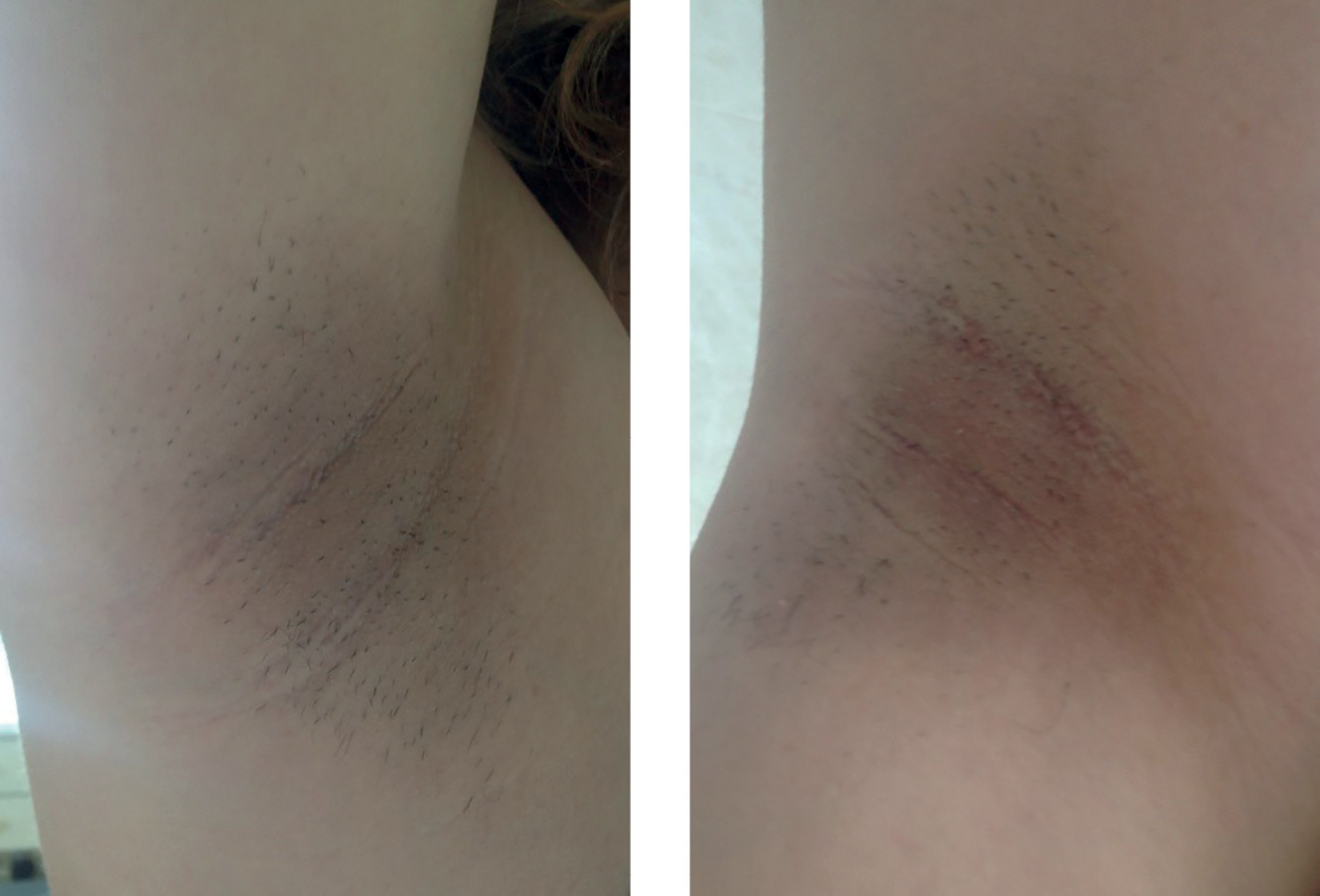
Рисунок 2. Acanthosis nigricans подмышечных впадин пациентки К. (впервые опубликовано Соркина Е.Л. и соавт.) [8].
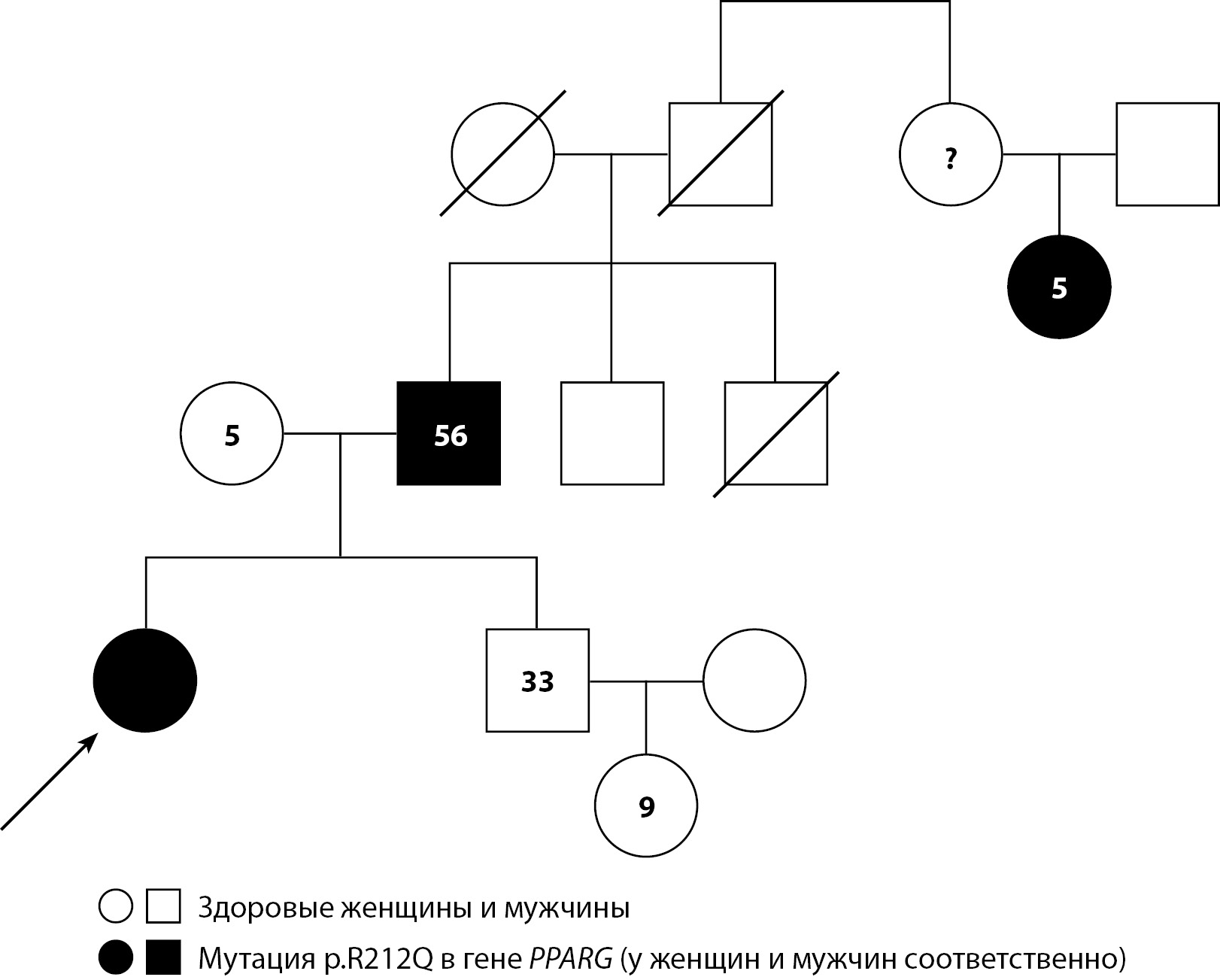
Рисунок 3. Генеалогическое древо пациентки К. (впервые опубликовано Соркина Е.Л. и соавт.) [8].
Пациентка Д., 51 г., с семейной парциальной липодистрофией 4 типа (патогенный вариант в гетерозиготном состоянии c.1210-1delG в гене PLIN1) и множественными метаболическими осложнениями поступила в отделение терапии сахарного диабета ФГБУ «НМИЦ эндокринологии» МЗ РФ в августе 2023 г. с жалобами на снижение чувствительности в нижних конечностях, отек левой стопы, появление пигментных пятен на голенях.
Из анамнеза известно, что с подросткового возраста наблюдались изменения внешности, характерные для парциальной липодистрофии: мускулистые верхние и нижние конечности, липогипертрофии в области лица и шеи.
При обследовании по поводу вторичной аменореи в возрасте 20 лет выявлена дислипидемия за счет крайне высоких уровней триглицеридов (до 43 ммоль/л), специализированного обследования не проходила.
В 2000 г. (28 лет) перенесла панкреонекроз. Впоследствии — неоднократные госпитализации с обострением хронического панкреатита с частотой 2–3 раза в год.
В 2011 г. (в 39 лет) диагностирован СД2, инициирована пероральная сахароснижающая терапия; назначались комбинации различных групп пероральных сахароснижающих препаратов, но компенсация углеводного обмена не была достигнута. В 2017 г. назначена комбинированная сахароснижающая терапия (инсулин гларгин — 300 Ед/мл 35 Ед утром, 40 Ед вечером, дапаглифлозин — 10 мг вечером, метформин — 850–1000 мг вечером).
В апреле 2021 г. с жалобами на давящие боли в левой половине грудной клетки, не связанными с физической нагрузкой, госпитализирована в ФГБУ «НМИЦ кардиологии», диагностирован 70% стеноз дистального сегмента огибающей артерии, 60% стеноз среднего и дистального сегмента артерии тупого края, окклюзия проксимального сегмента правой коронарной артерии. Учитывая малый диаметр огибающей артерии, хроническую окклюзию правой коронарной артерии, отсутствие типичных приступов стенокардии, рекомендована консервативная терапия (ацетилсалициловая кислота — 100 мг вечером, розувастатин — 40 мг, эзетимиб — 10 мг). В липидограмме отмечалось значительное повышение триглицеридов (до 33,31 ммоль/л), общего холестерина (8,75 ммоль/л), низкий уровень ЛПВП (0,66 ммоль/л).
В сентябре 2021 г. в связи с ранее выявленной гипертриглицеридемией инициирован плазмаферез 2 раза в месяц, на данный момент 1 раз в 3 недели.
Обращает на себя внимание семейный анамнез пациентки: мать — СД2, саркоидоз легких и внутригрудных лимфатических узлов; бабушка по материнской линии — СД2, бабушка по отцовской линии — ИБС. Фенотипически похожа на мать.
В ФГБУ «НМИЦ кардиологии» заподозрен диагноз семейной парциальной липодистрофии, рекомендовано генетическое исследование, которое было проведено в ФГБНУ «Медико-генетический научный центр имени академика Н.П. Бочкова» в марте 2023 г. В результате массового параллельного секвенирования кастомной панели из 60 генов был выявлен новый, ранее неописанный генетический вариант в гене перилипина PLIN1: NM_002666.5: c.1210-1delG в гетерозиготном состоянии. Наличие данного генетического варианта было подтверждено с помощью прямого автоматического секвенирования по Сэнгеру. На основании критериев ACMG (2015 г.) и руководства по интерпретации данных массового параллельного секвенирования [9] данный вариант был оценен как патогенный (критерии PM2, PVS1, PP5, PP4). После чего был выставлен диагноз: «Семейная парциальная липодистрофия из-за патогенного варианта в гене перилипина-1 (PLIN1)».
Длительное время отмечалось повышение показателей АД, максимально — до 210/100 мм рт.ст. Постоянно принимала эналаприл — 2,5 мг на ночь, затем самостоятельно отменила в связи с тенденцией к гипотонии. На данный момент адаптирована к показателям АД 120/70 мм рт.ст. на фоне приема лерканидипина 10 мг утром.
В 2017 г. (в 45 лет) при обследовании по поводу длительно непроходящего кашля выявлен саркоидоз легких и внутригрудных лимфатических узлов. По данным рентгенографии легких от 08.2023 г. (рис. 4), легкие умеренно эмфизематозны, корни расширены, фиброзны, бронхолегочный рисунок деформирован, усилен по интерстициально-сосудистому типу, имеется увеличение внутригрудных лимфатических узлов. Междолевая шварта справа. Со слов пациентки, она была консультирована пульмонологом, морфологическая верификация диагноза не требуется.
По данным УЗИ брюшной полости от 17.06.2023 г., визуализированы диффузные изменения паренхимы печени и поджелудочной железы; имеются эхографические признаки хронического панкреатита, дискинезии желчных путей.
В августе 2023 г. (51 г.) в связи с ухудшением общего самочувствия обратилась на консультацию в ФГБУ «НМИЦ эндокринологии», где была скорректирована сахароснижающая терапия: инсулин гларгин — 300 Ед/мл 80 Ед, дапаглифлозин — 10 мг вечером, метформин — 1000 мг утром и вечером, глимепирид —2 мг утром. Гликированный гемоглобин, со слов пациентки, от июня 2023 г. — 14%.
На момент госпитализации в 2023 г. получала: аторвастатин — 80 мг вечером, омакор — 1000 мг утром и вечером, эзетимиб — 10 мг вечером. Попытки назначения фенофибратов приводили к обострению панкреатита.
При осмотре пациентки отмечались: атлетическое телосложение, выраженный мышечный слой на голенях, бедрах, верхних конечностях, перераспределение ПЖК в область живота, лица и шеи (рис. 5). Масса тела 53,7 кг. Рост 160 см. ИМТ=21,0 кг/м2.
По данным проведенного обследования отмечалась декомпенсация углеводного обмена (HbA1c — 11,2%, гликемия в течение дня до 27,7 ммоль/л), в связи с чем отменен препарат сульфонилмочевины, инициирована инсулинотерапия инсулином аспарт + никотинамид + аргинин по 15–20 Ед перед основными приемами пищи, остальная терапия продолжена в прежнем объеме. Проведено индивидуальное обучение пациентки принципам интенсифицированной инсулинотерапии.
За время госпитализации проведена оценка наличия и выраженности поздних осложнений СД. Данных за наличие диабетической ретинопатии не получено. Установлена стадия поражения почек, микроальбуминурия (ХБП C1A2), подтверждено наличие диабетической дистальной полинейропатии.
Регулярно получает сеансы плазмафереза на фоне постоянной гиполипидемической терапии.
Проведен консилиум, по результатам которого, учитывая наличие тяжелого, прогрессирующего орфанного заболевания, высокий риск осложнений, в том числе острого панкреатита и сердечно-сосудистых событий, для коррекции метаболических нарушений рекомендовано в связи с жизненной необходимостью назначение терапии аналогом лептина — метрелептином.
Диагноз: «Сахарный диабет вследствие семейной парциальной липодистрофии 4 типа. Нефропатия сложного генеза (диабетическая, гипертоническая, нарушение пуринового обмена), ХБП С1А2. Дистальная диабетическая полинейропатия, сенсорная форма. ИБС: безболевая ишемия миокарда. Атеросклероз коронарных артерий (стеноз 70% дистального сегмента ОА, 60% стеноз артерии тупого края, хроническая окклюзия проксимального сегмента ПКА). Атеросклероз БЦА (стеноз ОСА справа 40%). Нестенозирующий атеросклероз артерий нижних конечностей. Гипертоническая болезнь III стадии, риск CCO4. Неалкогольная жировая болезнь печени. Дислипидемия. Хронический панкреатит, ремиссия. Вторичная аменорея. Мочекаменная болезнь, ремиссия. Саркоидоз легких и внутригрудных лимфатических узлов, без морфологической верификация, стадия регрессирования».
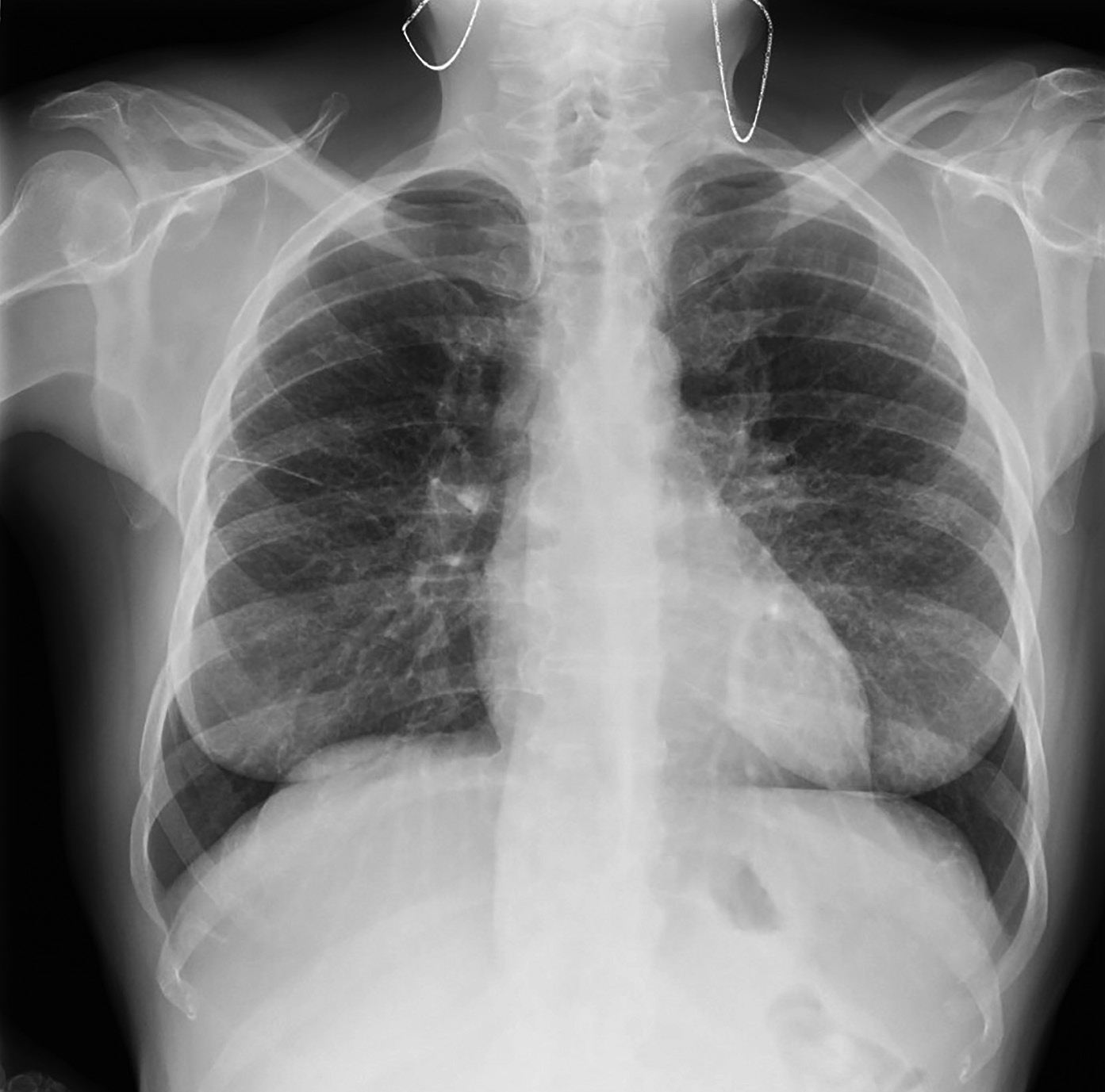
Рисунок 4. Рентенография легких пациентки Д.
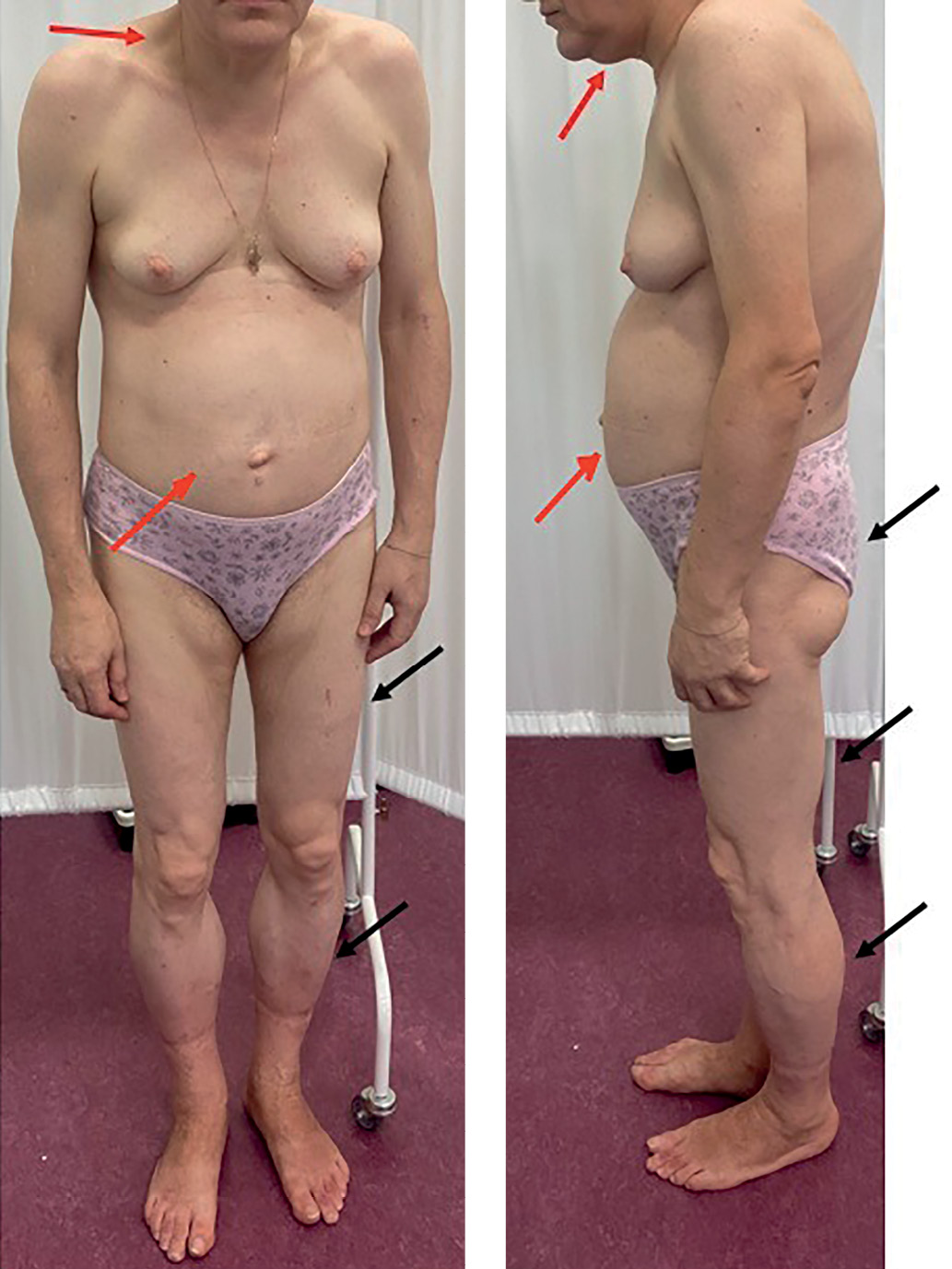
Рисунок 5. Фенотип пациентки Д. Красной стрелкой обозначены зоны липогипертрофии; черной — зоны липодистрофии.
Оба состояния (семейная парциальная липодистрофия и саркоидоз легких) являются редкими заболеваниями, а их сочетание встречается еще реже. Это заставляет задуматься о поиске общих патогенетических механизмов в развитии данных состояний.
Согласно исследованию, проведенному Е.Л. Соркиной в ФГБУ «НМИЦ эндокринологии» МЗ РФ, у многих пациентов с наследственными липодистрофиями отмечалась ассоциация с различными аутоиммунными заболеваниями: у 5 (41,7%) из 12 пациентов с врожденной генерализованной липодистрофией (ВГЛ) и у 3 (7,5%) из 40 с семейной парциальной липодистрофией (СПЛ) [11].
Misra A. и соавт. у 27 из 220 пациентов с СПЛ выявили связь с аутоиммунными заболеваниями [12]. Наиболее частым сопутствующим заболеванием была системная красная волчанка (СКВ), зарегистрированная у 7 пациентов [12]. Описаны три случая васкулитов, включая лейкоцитокластический васкулит и уртикарный васкулит и 4 случая дерматомиозита/полимиозита [12]. Кроме того, у 6 пациентов отмечалось повышение уровня антинуклеарного фактора в сыворотке крови без клинических признаков аутоиммунных заболеваний [12].
До настоящего времени остается актуальной проблема поиска этиологического фактора саркоидоза [13]. Существует несколько гипотез об этиологии этого заболевания, среди которых основными являются инфекционная и аутоиммунная [13].
Исследования геномных ассоциаций выявили наследственные факторы, влияющие на вероятность развития саркоидоза и на многообразие его клинических проявлений [14]. Во многих исследованиях определены гены — кандидаты восприимчивости к саркоидозу, например, гены интерлейкинов (IL1A, IL12B, IL18), HLA I и II класса, IFN-γ, CCR2, MST1R, BTNL2, CCR5, TNF-α, CCDC88B, XAF1, SLC11A2 или MST1) [14][15]. Повышенный риск развития саркоидоза связывают с мутациями в генах CFTR, OS9, ANXA11, KCNK4, NOTCH4, FAM117B, BAG2, SCGB1A1, RAB23, KIAA1586, RAGE, BEND6, PRDX5, RAS23VEGFA и ZNF415 [14]. Несмотря на многообразие перечисленных выше генов, среди них нет тех генов, мутации в которых ассоциированы с развитием семейных парциальных липодистрофий. У описанных нами пациентов исследование генов, ассоциированных с повышенным риском развития саркоидоза, не проводилось.
При анализе литературы за период с 2000 до 2023 гг. нет ни одного описания сочетания СПЛ и саркоидоза. Мы впервые приводим описание клинических случаев сочетания этих орфанных заболеваний. Согласно данным литературы, прямой связи между ними не описано. Необходимы дальнейшие исследования в области молекулярно-генетических основ развития наследственных липодистрофий, а также этиологии и патогенеза саркоидоза для выяснения связи между ними.
Источник финансирования. Работа выполнена за счет средств гранта Создание и развитие научного центра мирового уровня «Национальный центр персонализированной медицины эндокринных заболеваний» (соглашение № 075-15-2022-310).
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Участие авторов. Фролкова Н.В. — анализ медицинской документации пациента и написание текста; Кокшарова Е.О. — анализ литературных данных, окончательное утверждение для публикации рукописи; Васильев П.А. — анализ данных молекулярно-генетических анализов, редактирование текста; Смирнова О.М. — анализ литературных данных, окончательное утверждение для публикации рукописи; Шестакова М.В. — разработка концепции и дизайна, анализ литературных данных, окончательное редактирование текста и утверждение для публикации рукописи.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациентов. Авторы настоящей статьи получили письменное разрешение от упоминаемых в статье пациенток на публикацию их медицинских данных и фотографий в журнале «Сахарный диабет».
1. Соркина Е.Л., Тюльпаков А.Н. Наследственные и приобретенные липодистрофии: молекулярно-генетические и аутоиммунные механизмы // Ожирение и метаболизм. — 2018. — Т.15. — №1. — С. 39-42. doi: https://doi.org/10.14341/omet2018139-42
2. Patni N, Garg A. Lipodystrophy for the DiabetologistWhat to Look For. Curr Diab Rep. 2022;22(9):461-470. doi: https://doi.org/10.1007/s11892-022-01485-w
3. Hussain I, Patni N, Garg A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology. 2019;51(2):202-212. doi: https://doi.org/10.1016/j.pathol.2018.11.004
4. Балионис О.И., Никитин А.Г., Аверьянов А.В.Генетические предикторы течения саркоидоза легких в российской популяции // Вестник современной клинической медицины. — 2022. — №4. С. 18-25. doi: https://doi.org/10.20969/VSKM.2022.15(4).18-25
5. Starshinova AA, Malkova AM, Basantsova NY, et al. Sarcoidosis as an Autoimmune Disease. Front Immunol. 2020;10:2933. doi: https://doi.org/10.3389/fimmu.2019.02933
6. Spagnolo P, Maier LA. Genetics in sarcoidosis. Current Opinion in Pulmonary Medicine. 2021;27(5):423-429. doi: https://doi.org/10.1097/MCP.0000000000000798
7. Franzen DP, Brutsche M, Nilsson J, et al. Sarcoidosis - a multisystem disease. Swiss Med Wkly. 2022;152:w30049. doi: https://doi.org/10.4414/smw.2022.w30049
8. Соркина Е.Л., Калашникова М.Ф., Лиходей Н.В., и др. Развитие метаболического синдрома в молодом возрасте как проявление семейной парциальной липодистрофии 3 типа (дефект гена PPARG): первое описание клинического случая в России // Сахарный диабет. — 2015. — Т.18. — №3. — С. 99–105. doi: https://doi.org/10.14341/DM2015399-105
9. Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б., и соавт. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2) // Медицинская генетика. — 2019. — Т. 18. — №2. — С. 3-23
10. Fogacci F, Norata GD, Toth PP, Arca M, Cicero AFG. Efficacy and Safety of Volanesorsen (ISIS 304801): the Evidence from Phase 2 and 3 Clinical Trials. Curr Atheroscler Rep. 2020;22(5):18. doi: https://doi.org/10.1007/s11883-020-00836-w
11. Соркина Е.Л. Наследственные липодистрофии: клинические, гормональные и молекулярно-генетические характеристики: Дис. ... канд. мед. наук. — Москва; 2017. Доступно по: https://www.endocrincentr.ru/sites/default/files/specialists/science/dissertation/sorkinael_dissertacia.pdf. Ссылка активна на 13.06.2024.
12. Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore). 2004;83(1):18-34. doi: https://doi.org/10.1097/01.md.0000111061.69212.59
13. Starshinova AA, Malkova AM, Zinchenko YuS, et al. Autoimmune component in the etiology of sarcoidosis. Tuberculosis and Lung Diseases. 2020;98(5):54-62. (In Russ.) doi: https://doi.org/10.21292/2075-1230-2020-98-5-54-62
14. Яблонский П.К., Дробинцева А.О., Зубарева Т.С., и др. Саркоидоз: молекулярные маркеры и мишени таргетной диагностики и терапии // Молекулярная медицина. — 2022. — Т.20. — №3. — С. 3–10. doi: https://doi.org/10.29296/24999490-2022-03-01
15. Culver DA, Judson MA. New advances in the management of pulmonary sarcoidosis. BMJ. 2019;367:l5553. doi: https://doi.org/10.1136/bmj.l5553
Фролкова Надежда Викторовна, аспирант
117036, Москва, ул. Дм. Ульянова, д. 11
Кокшарова Екатерина Олеговна, н.с.
Москва
Васильев Петр Андреевич, н.с.
Scopus ID: 57202745394
WoS Researcher ID: AAN-4520-2020
Москва
Смирнова Ольга Михайловна, д.м.н., профессор, гл.н.с.
eLibrary SPIN: 9742-8875
Москва
Шестакова Марина Владимировна, д.м.н., профессор, академик РАН
Scopus Author ID: 7004195530
Москва
|
|
1. Рисунок 1. Фенотип пациентки К. Красной стрелкой обозначены зоны липогипертрофии; черной — зоны липодистрофии (впервые опубликовано Соркина Е.Л. и соавт.) [8]. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(207KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Acanthosis nigricans подмышечных впадин пациентки К. (впервые опубликовано Соркина Е.Л. и соавт.) [8]. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(117KB)
|
Метаданные ▾ | |
|
|
3. Рисунок 3. Генеалогическое древо пациентки К. (впервые опубликовано Соркина Е.Л. и соавт.) [8]. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(142KB)
|
Метаданные ▾ | |
|
|
4. Рисунок 4. Рентенография легких пациентки Д. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(205KB)
|
Метаданные ▾ | |
|
|
5. Рисунок 5. Фенотип пациентки Д. Красной стрелкой обозначены зоны липогипертрофии; черной — зоны липодистрофии. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(227KB)
|
Метаданные ▾ | |
Фролкова Н.В., Кокшарова Е.О., Васильев П.А., Смирнова О.М., Шестакова М.В. Сочетание семейной парциальной липодистрофии (синдром Даннигана - Кобберлинга) с саркоидозом легких. Сахарный диабет. 2024;27(3):287-294. https://doi.org/10.14341/DM13102
Frolkova N.V., Koksharova E.O., Vasiluev P.A., Smirnova O.M., Shestakova M.V. Combination of familial partial lipodystrophy (Dunnigan-Cobberling syndrome) with pulmonary sarcoidosis. Diabetes mellitus. 2024;27(3):287-294. (In Russ.) https://doi.org/10.14341/DM13102

 |
Адрес: 117036, Российская Федерация, Москва, улица Дмитрия Ульянова, дом 11.
Обработка персональных данных







































