




Содержание
Перейти к:
https://doi.org/10.14341/DM12819
Перейти к:
ОБОСНОВАНИЕ. Моногенный сахарный диабет (МСД) — это редкая форма сахарного диабета (СД), причиной которого является наличие одной или более мутаций в одном из генов, приводящих к дисфункции β-клеток поджелудочной железы. Несмотря на достаточную известность наиболее распространенных подтипов MODY, случаи МСД остаются недиагностированными и классифицируются как СД 1 и 2 типов.
ЦЕЛЬ. Изучить клинические, лабораторные характеристики, а также возрастные особенности GCK-MODY у детей.
МАТЕРИАЛЫ И МЕТОДЫ. Изучаемая популяция — пациенты с GCK-MODY в возрасте до 18 лет. Диагноз подтвержден результатом молекулярно-генетического исследования — выявление гетерозиготной мутации в гене GCK.
РЕЗУЛЬТАТЫ. MODY-GCK верифицирован у 144 пациентов (131 пробанда и 13 сибсов) в возрасте до 18 лет. В 80,2% случаев (n=105) были выявлены миссенс-мутации. В 59,6% случаев мутации выявлены однократно. Наиболее часто встречались миссенс-мутации p.G261R (n=7) и p.G258C (n=6). Возраст диагностики нарушений углеводного обмена составил 7,6 года [4,0; 11,2]. В 72,2% случаев нарушения углеводного обмена были диагностированы случайно, в 16,7% — обследование проведено по поводу отягощенной наследственности по СД, в 11,1% — отмечались клинические симптомы СД. Гликемия натощак при диагностике составила 6,8 ммоль/л [6,4; 7,3], уровень гликированного гемоглобина (HbA1c) — 6,4% [6,1; 6,7]. При обследовании уровень гликемии натощак соответствовал нормальным значениям у 16,4% пациентов, нарушенной гликемии натощак — у 57,8%, диабетическим значениям — 25,8%. На 120-й минуте при проведении орального глюкозотолерантного теста у 62,3% пациентов уровень гликемии соответствовал нарушенной толерантности к глюкозе, у 18,9% — диабетическим значениям, у 11,7% пациентов — нормальным показателям гликемии. Выявлена умеренная положительная корреляция между возрастом обследования и уровнями гликемии натощак (r=0,347; p<0,01), С-пептида (r=0,656; p<0,001) и инсулина (r=0,531; p<0,001). Инсулинорезистентность (ИР) по индексу НОМА выявлена у 21 пациента (14,5%), экзогенно-конституциональное ожирение — у 6 пациентов (4,2%). У 9 пациентов (6,25%) выявлено умеренное повышение титра специфических панкреатических антител (АТ). Наличие ИР, ожирения, АТ не оказывало влияния на уровень HbA1c. В 92,3% случаев компенсации углеводного обмена удавалось достичь на фоне диеты, в 4,2% был назначен инсулин, 2,1% — метформин, 1,4% — препараты сульфонилмочевины.
ЗАКЛЮЧЕНИЕ. У детей нарушения углеводного обмена при GCK-MODY диагностируются чаще всего случайно, асимптоматически в любом возрасте, в том числе с рождения, характеризуются сочетанием нарушения гликемии натощак и нарушения толерантности к глюкозе и, как правило, не требуют назначения сахароснижающей терапии.
Сечко Е.А., Кураева Т.Л., Зильберман Л.И., Лаптев Д.Н., Безлепкина О.Б., Петеркова В.А. Неиммунный сахарный диабет у детей, обусловленный гетерозиготными мутациями в гене глюкокиназы (GCK-MODY): анализ данных 144 пациентов. Сахарный диабет. 2022;25(2):145-154. https://doi.org/10.14341/DM12819
Sechko E.A., Kuraeva T.L., Zilberman L.I., Laptev D.N., Bezlepkina O.B., Peterkova V.A. Non-immune diabetes mellitus in children due to heterozygous mutations in the glucokinase gene (GCK-MODY): data of 144 patients. Diabetes mellitus. 2022;25(2):145-154. (In Russ.) https://doi.org/10.14341/DM12819
Моногенный сахарный диабет (МСД) — это редкая форма сахарного диабета (СД), причиной которого является наличие одной или более мутаций в одном из генов, приводящих к дисфункции β-клеток поджелудочной железы. Частота МСД достоверно не известна, по данным различных исследований, в детском возрасте его частота составляет 1–6% [1]. Наиболее распространенными формами МСД являются MODY (акроним названия maturity-onset diabetes of the young — диабет взрослого типа у молодых лиц). Термин MODY ввели R.B. Tattersall, S.S. Fajans в 1974–1975 гг., наблюдая 23 семьи с наследственной формой СД, в том числе семью Rinke-Wiegand, известную как семья RW, родословная которой состоит из более чем 360 членов семьи, при этом СД выявлен у 72 человек в 5 поколениях. В последующем в этой семье была выявлена гетерозиготная мутация в гене HNF4A Q268X [2][3]. Первоначально каждому подтипу MODY присваивался порядковый номер согласно очередности открытия генов: MODY1 — при гетерозиготной мутации в гене HNF4A, MODY2 — при мутациях в гене GCK и т.д. Известно 14 генов, приводящих к MODY [1]. В настоящее время от данной классификации постепенно отказываются, указывая в диагнозе непосредственно название гена: HNF4A-MODY, GCK-MODY и т.д.
Основные критерии, позволяющие заподозрить MODY: отягощенная наследственность по аутосомно-доминантного типу, отсутствие потребности или небольшая потребность в инсулине, отсутствие ожирения и инсулинорезистентности, а также отсутствие аутоантител (АТ), характерных для СД 1 типа (СД1) [1]. Золотым стандартом диагностики MODY является выявление гетерозиготных мутаций в соответствующих генах. По данным большинства авторов, в детском возрасте наиболее часто встречается GCK-MODY (MODY2), обусловленный инактивирующими гетерозиготными мутациями в гене глюкокиназы. Так, в Италии выявлено, что доля MODY среди всех форм СД у детей составляет 4,74%, причем преобладает GCK-MODY, который был верифицирован в 84,7% случаев MODY [4]. По данным W. Fendler, на долю GCK-MODY приходится 83% всех случаев MODY [5]. В исследовании А. Chakera et al. выявлена более высокая распространенность GCK-MODY — 1 на 1000 населения [6]. В России GCK-MODY встречается в 57,6–77,5% всех случаев MODY в детском возрасте [7–9].
Фермент глюкокиназа катализирует первую реакцию гликолитического метаболического пути — фосфорилирование глюкозы. Таким образом, глюкокиназа является связующим звеном между уровнем гликемии и началом секреции инсулина [10]. В настоящее время известно 679 мутаций в гене GCK, которые ассоциированы с фенотипом MODY (http://www.hgmd.cf.ac.uk). Не выявлено взаимосвязи между типом мутации, расположением мутации и степенью нарушения углеводного обмена [11].
Для GCK-MODY характерна доброкачественная, непрогрессирующая гипергликемия натощак, не требующая назначения сахароснижающей терапии [12]. Для сахарного диабета GCK-MODY также не характерно развитие микро- и макрососудистых осложнений. Приблизительно у 50% детей, у которых случайно выявлена гипергликемия, диагностирован GCK-MODY [13].
Несмотря на достаточную известность наиболее распространенных подтипов MODY, случаи моногенного диабета остаются недиагностированными и классифицируются как СД1 и СД 2 типа (СД2). По данным исследования, проведенного в рамках SEARCH, в 94% случаев среди подростков и детей наиболее распространенные формы МСД диагностировались как СД1 и СД2, и в 76% случаев данные пациенты получали терапию, которая была им не показана [14]. Все это свидетельствует о необходимости дальнейшего изучения клинических характеристик GCK-MODY, что будет способствовать совершенствованию дифференциальных различных форм СД и выявлению новых случаев МСД.
Диагноз GCK-MODY позволяет прогнозировать течение заболевания, проводить генетическое консультирование родственников пациента, а также отменить получаемую пациентом сахароснижающую терапию, в том числе инсулин, что снижает инвазивность терапии, повышает качество жизни пациентов, а также снижает затраты на лечение.
Изучить клинические, лабораторные характеристики, а также возрастные особенности GCK-MODY у детей.
Место и время проведения исследования
Место проведения. Исследование проведено в ФГБУ «НМИЦ эндокринологии» Минздрава России, в детском отделении сахарного диабета.
Время исследования. Исследование проводилось с сентября 2013 г. по декабрь 2020 г.
Изучаемые популяции (одна или несколько)
Изучаемая популяция — пациенты с GCK-MODY в возрасте до 18 лет.
Критерием включения в исследование являлось выявление гетерозиготной мутации в гене GCK. Критериями направления на молекулярно-генетическое исследование были возраст менее 18 лет, отсутствие или небольшая потребность в инсулине (менее 0,5 Ед/кг/сут), сохранная секреция инсулина и С-пептида, отсутствие специфических панкреатических аутоантител в анамнезе и/или отягощенный семейный анамнез по СД.
Способ формирования выборки из изучаемой популяции (или нескольких выборок из нескольких изучаемых популяций)
Выборка формировалась сплошным методом — в исследование включены пациенты с диагнозом GCK-MODY, который был установлен при выявлении гетерозиготных мутаций в гене GCK (131 пробанд и 13 сибсов).
Дизайн исследования
Исследование одноцентровое наблюдательное одномоментное одновыборочное.
Методы
Молекулярно-генетическое исследование проводилось методом прямого секвенирования экзонов 1а, 2–10 и примыкающих участков интронов гена GCK или методом NGS [15]. Определялись АТ к цитоплазматическим структурам β-клеток (ICA), глутаматдекарбоксилазе (GADA), тирозинфосфатазе (IA-2) и антиинсулиновые АТ (IAA), АТ к транспортеру цинка 8 (ZnT8 Ab). Изучение клинических характеристик включало: анализ жалоб, анамнеза, семейного анамнеза, оценку антропометрических показателей. ИМТ (кг/м2) рассчитывался как отношение массы тела (кг) к квадрату длины тела (м²), оценивался по нормативам ВОЗ для конкретного возраста и пола и был представлен в виде числа стандартных отклонений от среднего (SDS — standard deviation score). За диагностический критерий ожирения был принят SDS ИМТ>2,0 (ВОЗ, 2007).
Были изучены клинико-лабораторные характеристики обследованных пациентов: уровень гликированного гемоглобина (HbA1c), уровни глюкозы, С-пептида, инсулина. Оценка состояния углеводного обмена, секреции инсулина и С-пептида проводилась при проведении стандартного орального глюкозотолерантного теста (ОГТТ). Для оценки инсулинорезистентности (ИР) рассчитан индекс HOMA-IR (homeostasis model assessment) по формуле:
(ИРИ0×Гл0)/22,5,
где ИРИ — иммунореактивный инсулин, мкЕд/мл; Гл — глюкоза, ммоль/л.
ИР диагностировалась при значении индекса НОМА>3,2.
Статистический анализ
Статистическая обработка полученных данных проводилась с использованием пакета статистических программ IBM SPSS Statistics 26.0 (США). Данные представлены в виде медианы значения и интерквартильного размаха (Ме [25; 75 перцентиль]). Для сравнения двух независимых выборок по количественным признакам использовался критерий Манна–Уитни, по качественным признакам — критерий хи-квадрат (χ2). Для анализа связи двух признаков использовался анализ ранговой корреляции по Спирмену. Критический уровень значимости различий принимался при p<0,05.
Этическая экспертиза
Протокол исследования был одобрен на заседании локального этического комитета ФГБУ «НМИЦ эндокринологии» Минздрава России (протокол №7 от 24.04.2019 г.).
Молекулярно-генетическое исследование гена GCK была проведено 312 пациентам, диагноз MODY-GCK верифицирован у 144 пациентов (131 пробанда и 13 сибсов) в возрасте до 18 лет (46,1% случаев). Соотношение девочек к мальчикам 1:1,25.
При одноплодной беременности при сроке гестации 38–41 нед длина тела при рождении составила 51,5 см [-0,51; 1,24] (SDS 0,62 [-0,51; 1,24]), масса тела — 3145 г [ 2800; 3453] (SDS -0,63 [-1,7; 0,1]).
Особенности диагностики нарушений углеводного обмена (табл. 1). Медиана возраста диагностики нарушений углеводного обмена составила 7,6 года [ 4,0; 11,2]. Нарушения углеводного обмена в 72,2% были диагностированы случайно (n=104), в 16,7% (n=24) — обследование проведено по поводу отягощенной наследственности по СД, в 11,1% (n=13) — отмечались клинические симптомы СД (полидипсия (n=13), полиурия (n=10), снижение массы тела на 1–3 кг (n=6), глюкозурия (n=4), кетонурия (n=4)). Медиана уровня гликемии натощак при диагностике составила 6,8 ммоль/л [ 6,4; 7,3], HbA1c — 6,4% [ 6,1; 6,7].
Таблица 1. Клинико-лабораторная характеристика пациентов с GCK-MODY при диагностике нарушений углеводного обмена
Клинико-лабораторный показатель | Значение |
Соотношение полов, ж : м | 1:1,25 |
Возраст диагностики, лет | 7,6 [ 4,0; 11,2] |
Гликемия натощак при диагностике, ммоль/л | 6,8 [ 6,4; 7,3] |
HbA1c при диагностике, % | 6,4% [ 6,1; 6,7] |
Отягощенная наследственность, %: нарушения углеводного обмена у родителей нарушения углеводного обмена в 3 поколениях | 79,4 50,4 |
Характер диагностики, %: случайная обследование по поводу отягощенной наследственности клинические проявления СД | 72,2 16,7 11,1 |
Терапия, %: диета инсулин метформин препараты сульфонилмочевины | 90,9 4,2 4,2 0,7 |
Особенности диагностики GCK-MODY до 6 мес жизни. У 9 пациентов (6,25%) нарушения углеводного обмена были диагностированы в возрасте до 6 мес, медиана возраста диагностики составила 0,3 года [ 0,1; 0,5], минимальный возраст диагностики — 2 дня жизни, то есть GCK-MODY может входить в структуру неонатального СД. У большинства пациентов (n=8) нарушения углеводного обмена выявлены случайно при рутинном обследовании. Один пациент обследован в связи с выявленной глюкозурией. Показатели углеводного обмена не различались у пациентов при диагностике до 6 мес жизни и старше (n=136): гликемия — 6,8 ммоль/л [ 6,4; 7,0] и 6,8 ммоль/л [ 6,4; 7,3] соответственно, уровень HbA1c — 6,4% [ 5,7; 6,9] и 6,4% [ 6,1; 6,7]. Сахароснижающая терапия не была назначена ни одному пациенту.
Данные обследования при молекулярно-генетической верификации GCK-MODY (табл. 2). Возраст на момент молекулярно-генетической верификации диагноза составил 10,3 года [ 7,4; 15,0], длительность заболевания — 1,9 года [ 0,7; 3,7]. ИМТ 17,8 кг/м2 [ 15,4; 20,3], SDS ИМТ 0 [ -0,6; 0,7]. Уровень HbA1c 6,5% [ 6,2; 6,7]. Диагностический уровень HbA1c определялся у 50,5% пациентов. При проведении ОГТТ (n=85) уровень гликемии натощак составил 6,5 ммоль/л [ 6,1; 7,0], на 60-й минуте — 10,5 ммоль/л [ 8,9; 12,2], на 120-й минуте — 9,1 ммоль/л [ 8,0; 10,3].
Таблица 2. Клинико-лабораторная характеристика пациентов с GCK-MODY при верификации диагноза
Клинико-лабораторный показатель | Значение |
Возраст обследования, лет | 10,3 [ 7,4; 15,0] |
Длительность заболевания, лет | 1,9 [ 0,7; 3,7] |
HbA1c, % | 6,5 [ 6,2; 6,7] |
ИМТ, кг/м2 | 17,8 [ 15,4; 20,3] |
SDS ИМТ | 0 [ -0,6; 0,7] |
Частота ожирения, % | 4,2 |
Частота инсулинорезистентности, % | 14,5 |
Уровень гликемии натощак соответствовал нормальным значениям у 16,4% пациентов, нарушенной гликемии натощак (НГН) — у 57,8%, диабетическим значениям — 25,8%. На 120-й минуте при проведении ОГТТ у 62,3% пациентов уровень гликемии соответствовал нарушенной толерантности к глюкозе (НТГ), у 18,9% — диабетическим значениям, у 11,7% пациентов на 120-й минуте определялся нормальный уровень гликемии. У 77,1% пациентов хотя бы один из показателей (гликемии натощак, стимулированного уровня гликемии на 120-й минуте в ходе ОГТТ, уровень HbA1c) достигал диабетических значений. Выявлена умеренная положительная корреляция между уровнем гликемии натощак и возрастом пациентов при обследовании (r=0,347; p<0,01) (рис. 1). Уровень HbA1c не зависел от возраста обследования (r=0,208; p>0,05).
Уровень С-пептида натощак составил 1,5 нг/мл [ 1,0; 1,9], на 60-й минуте — 5,3 нг/мл [ 4,1; 7,1], на 120-й минуте — 4,9 нг/мл [ 3,7; 6,7]. Уровень инсулина натощак 6,2 мкЕд/мл [ 3,6; 9,0], на 60-й минуте — 38,2 мкЕд/мл [ 23,1; 54,1], на 120-й минуте — 30,2 мкЕд/мл [ 17,0; 48,2]. Выявлена умеренная положительная корреляция между возрастом обследования и уровнями С-пептида (r=0,656; p<0,001) (рис. 2) и инсулина (r=0,531; p<0,001) (рис. 3).
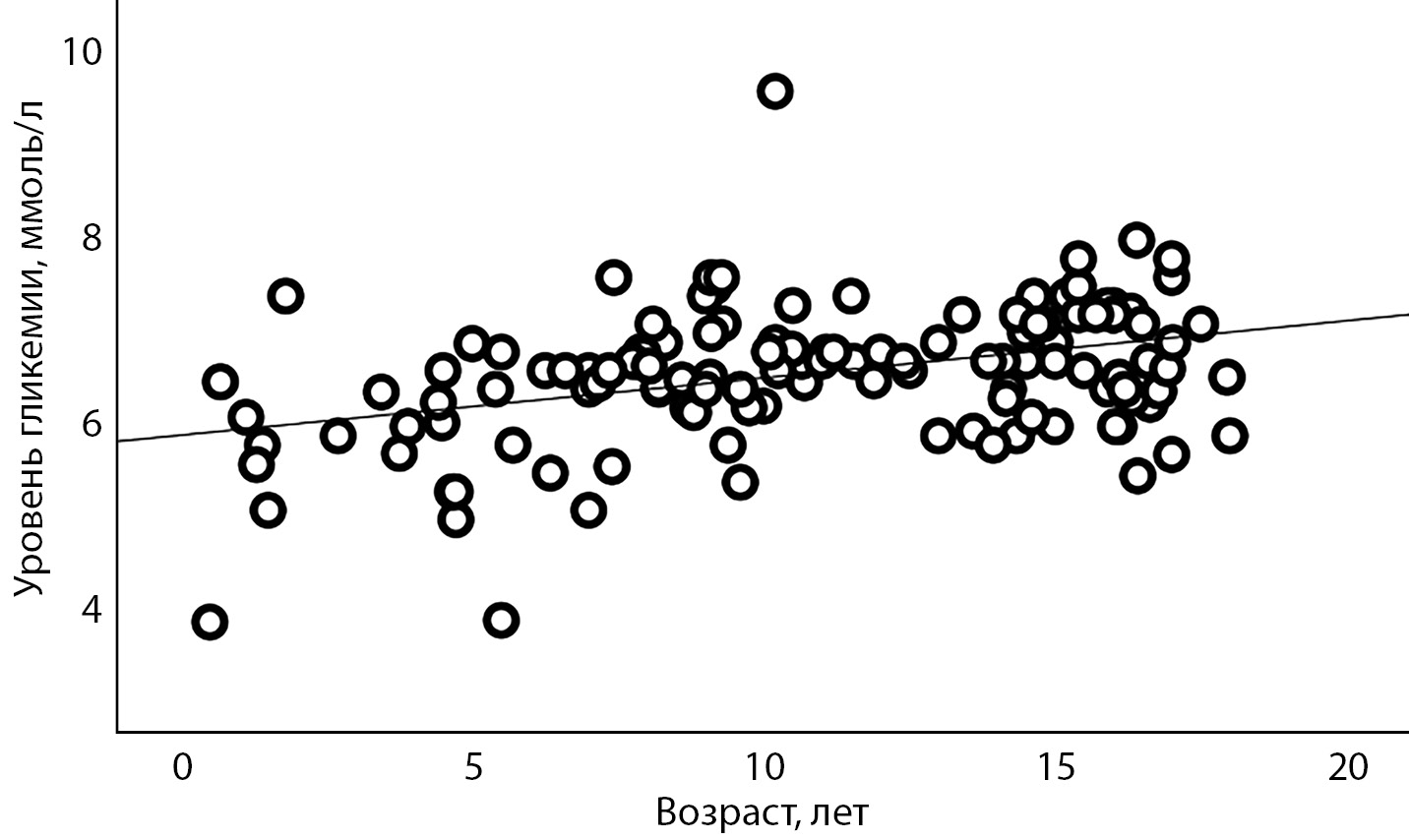
Рисунок 1. Корреляционная взаимосвязь между возрастом обследования и уровнем гликемии натощак у детей с GCK-MODY.
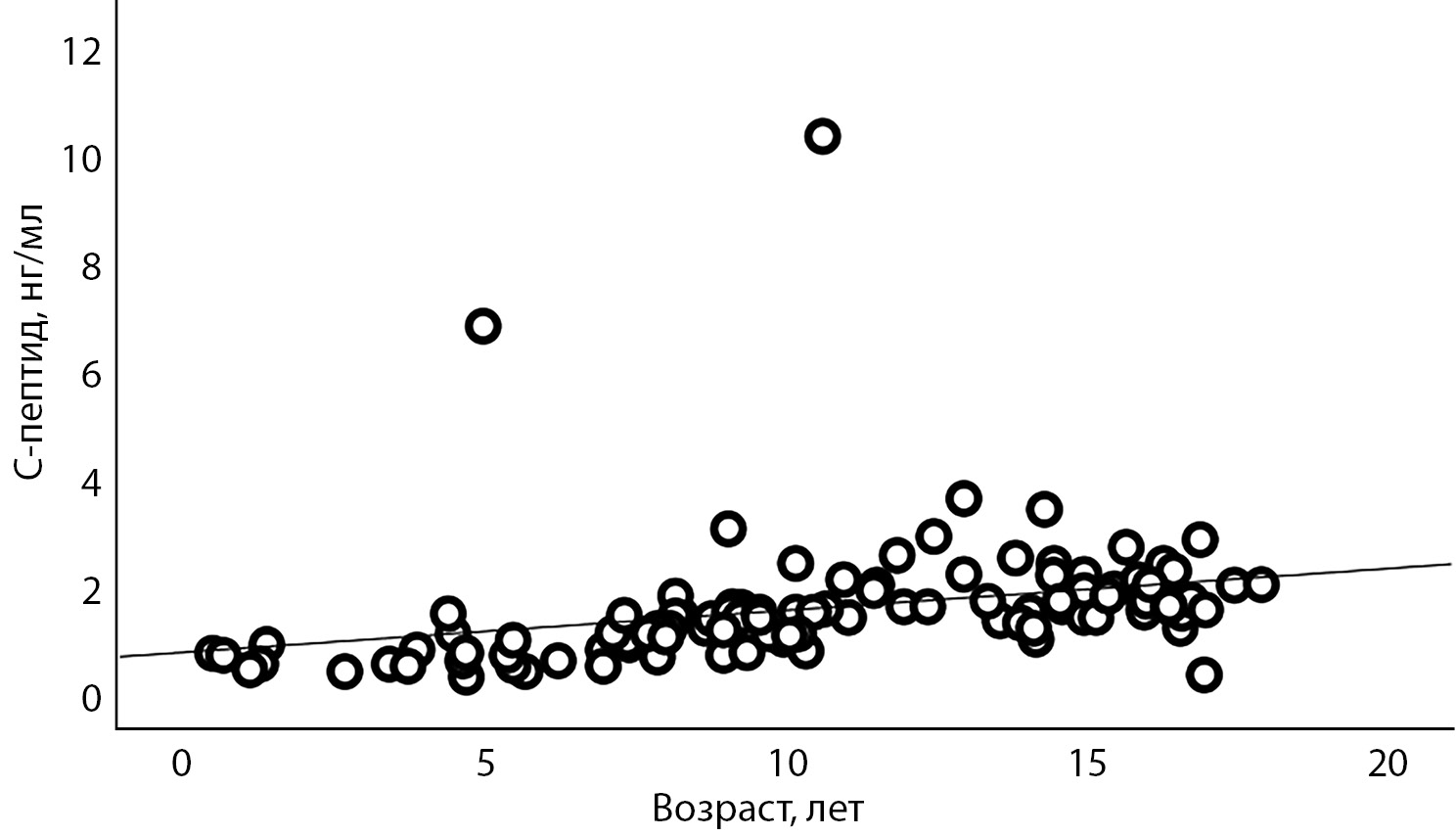
Рисунок 2. Корреляционная взаимосвязь между возрастом обследования и уровнем С-пептида натощак у детей с GCK-MODY.
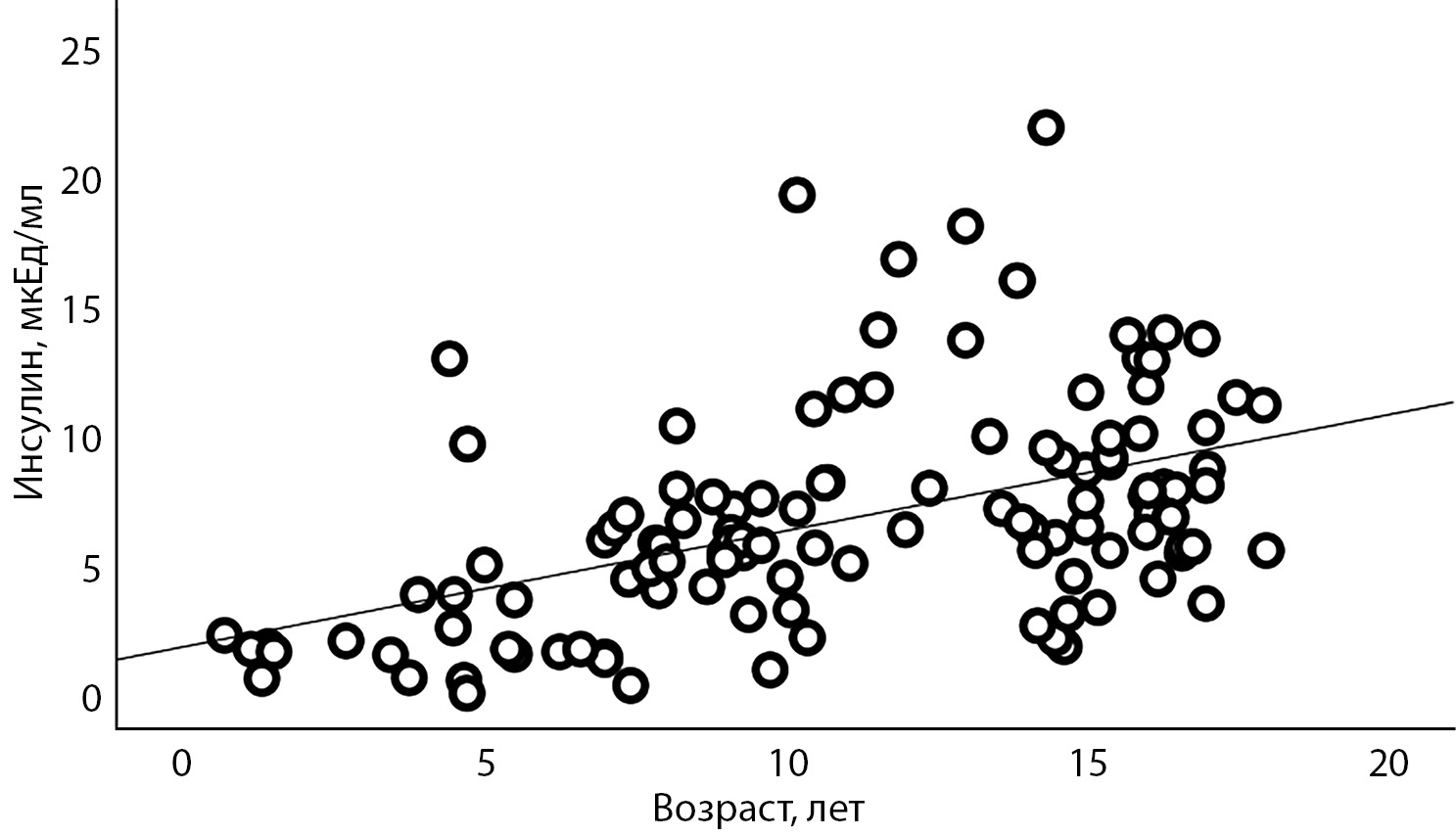
Рисунок 3. Корреляционная взаимосвязь между возрастом обследования и уровнем инсулина натощак у детей с GCK-MODY.
ИР по индексу НОМА выявлена у 21 пациентов (14,5%) — медиана индекса HOMA 1,8 [ 1,1; 2,8] при норме менее 3,2. Пациенты с выявленной ИР были старше пациентов без ИР — 14 лет [ 11,5; 16,1] против 9,9 года [ 7,2; 15,0], p>0,05. SDS ИМТ достоверно не отличалось 0,5 [-0,5; 1,2] и -0,2 [-0,8; 0,6], p>0,05. Показатели углеводного обмена в 2 группах не различались: уровень HbA1c составлял 6,5% [ 6,3; 6,9] и 6,4% [ 6,2; 6,7], глюкоза натощак — 6,7 ммоль/л [ 6,4; 7,3] и 6,5 ммоль/л [ 6,0; 6,9], на 120-й минуте при ОГТТ — 9,0 ммоль/л [ 7,3; 10,8] и 9,2 ммоль/л [ 8,2; 10,4] у пациентов с ИР и без ИР соответственно. Экзогенно-конституциональное ожирение отмечалось у 6 пациентов (4,2%). SDS ИМТ не коррелировало с возрастом диагностики (r=0,19; p>0,05) и уровнем HbA1c (r=-0,11; p>0,05).
Терапия GCK-MODY. При диагностике нарушений углеводного обмена в 91% случаев рекомендована диета (n=131), в 4,2% (n=6) был назначен инсулин в дозе 0,05–0,4 еД/кг/сут, в 4,2% — метформин (n=6) в дозе 500–2500 мг/сут, в одном случае — глибенкламид 1,75 мг/сут. До молекулярно-генетической верификации диагноза MODY 15,4% пациентов получали сахароснижающую терапию: 7,7% — инсулин (n=11), 5,6% — метформин (n=8), 2,1% — препараты сульфонилмочевины. Диагностика GCK-MODY позволила снизить долю пациентов, получающих сахароснижающую терапию, до 7,7%: 4,2% (n=6) — получали инсулин, 2,1% (n=3) — метформин, 1,4% (n=2) — препараты сульфонилмочевины.
Наследственный анамнез. Наследственность по СД отягощена у 88,5% пробандов (n=116), в том числе в 3 поколениях — у 50,4% (n=66). Нарушения углеводного обмена были выявлены у 79,4% (n=104) родителей пробандов (59,7% — у матери (n=62), 40,3% — у отца (n=42)), из них СД диагностирован у 40,3% (n=42), гестационный СД — у 18,2% (n=19), НГН и НТГ — у 33,6% (n=35). В 8 случаях нарушения углеводного обмена у одного из родителей были выявлены при активном обследовании в связи с подозрением на наследственную форму СД у ребенка. Возраст диагностики нарушений углеводного обмена у родителей составил 31 год [27,0; 38,0]. В большинстве случаев родители пробандов с нарушениями углеводного обмена (76,9%, n=80) сахароснижающую терапию не получали. В 20,2% (n=21) родители пробандов получали пероральные сахароснижающие препараты (ПССП), в 2,9% (n=3) — инсулин.
Исследование специфических панкреатических аутоантител. У 9 пациентов определялись АТ в невысоком титре (табл. 3): у 8 пациентов определялось повышение одного типа АТ, у одной пациентки выявлено повышение двух типов АТ (ZnT8a и GAD). Не выявлено связи между наличием антител и уровнем HbA1c (6,6% [ 6,5; 6,7] у пациентов с АТ и 6,5% [ 6,3; 6,7], p>0,05) при одинаковой длительности заболевания (2,0 года [ 1,1; 3,7] у пациентов с АТ и 2,0 года [ 0,7; 3,7] у пациентов без АТ, p>0,05). Пациенты с повышением титра АТ в терапии не нуждались.
Молекулярно-генетическое исследование. У пробандов в большинстве случаев были выявлены миссенс-мутации — 80,2% (n=105), в 7,6% (n=10) — делеции и инсерции со сдвигом рамки считывания, в 6,8% (n=9) — нонсенс-мутации, в 5,3% (n=7) — мутации в интроне. В 59,6% мутации выявлены однократно. Мутации, выявленные у нескольких неродственных пробандов, представлены в табл. 4. У одной пациентки выявлено две мутации в гене GCK — ранее описанная p.M462I [16] и неописанная p.410F.
Таблица 3. Специфические панкреатические антитела у детей с MODY-GCK
Вид АТ | Титр АТ, Ед/мл | Норма | Число (%) АТ+ | Число обследованных пациентов |
IA2 | 27–28 | 0–10 | 3 | 74 |
IAA | 14–21 | 0–10 | 3 | 94 |
GAD | 10 | 0–10 | 1 | 112 |
ZnT8a | 18,4–20 | 0–15 | 2 | 20 |
ICA | 20 | 0–10 | 1 | 103 |
Таблица 4. Частые мутации в гене GCK у пробандов
Мутация | Тип мутации | Описана/не описана | Количество пробандов |
p.R36W | Миссенс | [17] | 3 |
p.G44S | Миссенс | [18] | 4 |
p.G72R | Миссенс | [19] | 3 |
p.F150Y | Миссенс | [20] | 5 |
p.A188T | Миссенс | [21] | 2 |
p.R191W | Миссенс | [22] | 4 |
p.T206M | Миссенс | [23] | 2 |
p.E256K | Миссенс | [24] | 4 |
p.G258C | Миссенс | [25] | 6 |
p.G261R | Миссенс | [26] | 7 |
p.E265K | Миссенс | [27] | 2 |
p.Y273N | Миссенс | Не описана | 2 |
p.L324P | Миссенс | [28] | 2 |
p.C372X | Нонсенс | Не описана | 2 |
p.S383L | Миссенс | [29] | 3 |
int6+2T>G | Мутация в интроне | Не описана | 2 |
В нашем исследовании проанализирована большая группа пациентов в возрасте до 18 лет с GCK-MODY — 144 человека. Как известно, в детском возрасте GCK-MODY (MODY2) является наиболее распространенным подтипом MODY: в Польше на долю GCK-MODY приходится 83% случаев моногенного СД в детском возрасте, в Италии — 84,7%, в Германии и Австрии — 62% [4][5]. В РФ, по данным различных исследований, также отмечается преобладание GCK-MODY [7–9]. Причиной персистирующей гипергликемии натощак у детей в 50% являются мутации в гене глюкокиназы [13]. По данным исследования А. Chakera и соавт., проведенном у женщин с гестационным СД, распространенность GCK-MODY составляет 1:1000 [6].
Ген глюкокиназы локализован на 12 хромосоме и состоит из 12 экзонов, кодирует 465 аминокислот, экспрессируется в поджелудочной железе, печени, головном мозге, эндокринных клетках ЖКТ [30]. Глюкокиназа играет важнейшую роль в регуляции секреции инсулина, она является связующим звеном между уровнем гликемии и началом секреции инсулина, поэтому ее называют сенсором глюкозы в β-клетках поджелудочной железы. Регуляция секреции инсулина осуществляется благодаря его кинетике — скорость фосфорилирования глюкозы изменяется пропорционально ее концентрации в крови [10].
Мутации в гене GCK ассоциированы с двумя фенотипами СД. Перманентный неонатальный СД развивается при гомозиготных или компаундных гетерозиготных инактивирующих мутациях в гене GCK [31]. При инактивирующих гетерозиготных мутациях наблюдается фенотип GCK-MODY. При инактивирующих гетерозиготных мутациях в гене GCK фосфорилирование глюкозы и, следовательно, секреция инсулина происходят при более высоких показателях гликемии. Также у пациентов с GCK-MODY отмечаются снижение продукции гликогена в печени и усиленный глюконеогенез после еды. При проведении эугликемического клэмпа отмечено снижение супрессии глюконеогенеза в печени при физиологической концентрации инсулина [32]. Таким образом, гомеостаз глюкозы осуществляется при более высоких значениях гликемии, хотя регуляция секреции инсулина не нарушена, что клинически проявляется в виде стабильной гипергликемии. Клиническое течение GCK-MODY — наиболее мягкое из всех подтипов MODY и в связи с наибольшей распространенностью определяет отношение к MODY как легкой форме диабете.
В нашем исследовании нарушения углеводного обмена у детей с GCK-MODY в 88,9% носили асимптоматический характер и были диагностированы случайно или при обследовании в связи с отягощенной наследственностью по СД. Лишь у 13 пациентов отмечались клинические симптомы СД. В том числе у 4 пациентов поводом для исследования углеводного обмена послужила выявленная кетонурия, при обследовании гликемия составляла 5,6–9,0 ммоль/л, уровень HbA1c — 5,3–6,8%. В то же время кетоацидоз не характерен для MODY, более того, он является критерием исключения MODY [1]. Мы не нашли в доступной литературе описания случаев кетонурии или кетоацидоза при GCK-MODY, вероятно, кетонурия у детей в нашем исследовании не обусловлена нарушениями углеводного обмена, а является проявлением ацетонемического синдрома у детей. Тем не менее наличие кетонурии при мягкой гипергликемии не должно быть основанием для исключения MODY. Глюкозурия не характерна для GCK-MODY, но была выявлена у 4 детей при диагностике нарушений углеводного обмена, при обследовании гликемия составляла 6,6–8,0 ммоль/л, уровень HbA1c — 5,9–6,7%. Глюкозурия в других исследованиях также выявлена в 6,5% случаев [33].
Возраст диагностики нарушений углеводного обмена варьировал в широком диапазоне — от 2 дней жизни до 16,5 года. У 9 пациентов (6,25%) дебют нарушений углеводного обмена был в возрасте до 6 мес, таким образом, MODY-GCK может входить в структуру неонатального СД. Однако даже при столь ранней диагностике нарушения углеводного обмена носили асимптоматический характер и не требовали назначения сахароснижающей терапии. A. Huges et al. описали три клинических случая неонатальной гипергликемии, обусловленной гетерозиготными мутациями в гене GCK. И.И. Дедов и соавт. также описали три случая диагностики GCK-MODY в возрасте до 6 мес. Во всех случаях гипергликемия была выявлена случайно в возрасте от 1 сут жизни до 6 мес, сахароснижающая терапия не назначалась [34][35]. Таким образом, по возрасту диагностики нарушений углеводного обмена GCK-MODY может классифицироваться как неонатальный СД, однако данные нарушения имеют доброкачественный характер и не требуют назначения инсулина.
Для GCK-MODY характерен достаточно специфический фенотип — гипергликемия натощак в сочетании с НТГ на 120-й минуте при проведении ОГТТ. В нашем исследовании у детей в 83,6% случаев были выявлены НГН или диабетический уровень гликемии натощак. При проведении ОГТТ у 62,3% пациентов была выявлена НТГ. У 22,9% детей с мутацией в гене GCK ни один из показателей углеводного обмена не достигал диагностических критериев СД. При изучении GCK-MODY среди взрослых лиц отмечается прогрессирование уровня HbA1c по мере увеличения возраста (0,2 ммоль/моль в год), которое, однако, оказалось сопоставимо (p=0,06) с прогрессированием уровня HbA1c в группе контроля (здоровые члены семей) [36]. Нами выявлена умеренная положительная корреляция между уровнем гликемии натощак и возрастом пациентов, однако не выявлено повышения уровня HbA1c с увеличением возраста обследования. Таким образом, нельзя однозначно говорить об отсутствии прогрессирования нарушений углеводного обмена в детском возрасте при GCK-MODY.
В нашем исследовании экзогенно-конституциональное ожирение было выявлено у 4,2% детей, что сопоставимо (p>0,05) с данными мультицентрового исследования (Астрахань, Екатеринбург, Красноярск, Самара, Санкт-Петербург) по изучению частоты ожирения среди детского населения, в котором распространенность ожирения составила 5,7% [37]. Для MODY ожирение не характерно, однако с наблюдающимся распространением ожирения в детском возрасте любая форма СД может сочетаться с ожирением, приводя к гипердиагностике СД2 у детей. В исследовании TODAY у 4,5% детей с СД2 диагностирован MODY, из них у 31,8% — GCK-MODY [38]. По данным исследований SEARCH, TODAY у детей с ожирением при отсутствии АТ необходимо проведение молекулярно-генетического исследования для исключения GCK-MODY [14][38].
Вопрос ИР при GCK также остается открытым. Ожирение и ассоциированная с ожирением ИР обсуждаются в качестве фактора более ранней диагностики всех типов СД, в том числе и MODY [39][40]. Мы не получили данных о более ранней диагностике нарушений углеводного обмена у пациентов с GCK-MODY с ИР по сравнению с пациентами без ИР. Наличие ИР у пациентов не оказывало влияния на показатели углеводного обмена. Однако пациенты с ИР были значимо старше пациентов без ИР, также была выявлена положительная корреляция между возрастом обследования и индексом HOMA. Более низкая чувствительность к инсулину при GCK-MODY у детей в возрасте 13–18 лет по сравнению с допубертатными детьми и лицами старше 18 лет выявлена в исследовании K. Clément. Авторы объясняют сниженную чувствительность к инсулину пубертатной ИР [41]. Эти данные позволяют предполагать, что в развитие ИР у детей с мутацией в гене GCK наибольший вклад вносит пубертатная ИР, которая отмечается у детей с нормальной массой тела и без нарушений углеводного обмена [42].
Высокая концентрация СД в семье (отягощенный анамнез по СД в 3 поколениях) является одним из ключевых критериев, позволяющих заподозрить моногенную форму заболевания. Однако в нашем исследовании только у 50,4% пробандов наследственность была отягощена в 3 поколениях, что, вероятнее всего, обусловлено недостаточной осведомленностью пациентов о состоянии здоровья родственников 2-й степени родства, а также в связи с мягкостью клинических проявлений GCK-MODY. У 8 родителей нарушения углеводного обмена были выявлены активно в связи с подозрением на наличие моногенной формы СД у ребенка. У 21,6% родителей нарушений углеводного обмена не установлено. Это может быть обусловлено недостаточной информированностью о состоянии здоровья одного из родителей по социальным причинам (развод, смерть одного из родителей и т.д.). Аутосомно-доминантное наследование не будет также прослеживаться при мутациях de novo. В исследовании в двух национальных центрах Словакии и Чехии проведено молекулярно-генетическое исследование генов GCK, HNF1a, HNF4a у 150 пробандов без АТ, выраженного ожирения и отягощенной наследственности. В изучаемой когорте пациентов диагноз MODY был подтвержден в 39% случаев (58 пробандов), в том числе GCK-MODY — в 29,3% (44 пробандов), из них в 18 случаях имелось бессимптомное носительство мутации родителей, в 6 случаях — мутации de novo [43].
GCK-MODY — это неиммунная форма СД, однако в нашем исследовании мы не исключали пациентов с повышением титра АТ при наличии у них отягощенной наследственности по СД, что позволило выявить мутации в гене GCK у 9 пациентов с АТ. АТ определялись в невысоком титре, превышающем верхнюю границу нормы не более чем в 2 раза. Наличие АТ не оказывало влияния на течение заболевания — сахароснижающая терапия не была назначена ни одному пациенту, уровень HbA1c при одинаковой длительности заболевания был сопоставим с уровнем HbA1c у пациентов без АТ. Однако требуется дальнейшее наблюдение за пациентами с GCK-MODY с АТ для оценки влияния наличия АТ на прогрессирование нарушений углеводного обмена.
В гене GCK не выявлено частых мутаций, в основном мутации (59%) встречаются в одной семье каждая [16]. В нашем исследовании также в большинстве случаев (59,6%) мутации встречались однократно. Наиболее часто встречались миссенс-мутации p.G261R и p.G258C, выявленные у 7 и 6 неродственных пробандов соответственно.
Во многих исследованиях показано, что пациентам с GCK-MODY для компенсации углеводного обмена достаточно диетотерапии, за исключением случаев гестационного СД, обусловленного мутациями в гене GCK, когда показана терапия инсулином при риске макросомии плода [44]. Более того, показано, что сахароснижающая терапия при GCK-MODY не приводит к улучшению показателей углеводного обмена. Предполагается, что отсутствие эффективности терапии тесно связано с особенностями регуляции углеводного обмена при GCK-MODY, который осуществляется на более высоких показателях гликемии, чем у здоровых людей. Сниженный синтез гликогена в печени, повышенный глюконеогенез после еды, а также ранний контррегуляторный ответ на гипогликемию противодействуют сахароснижающему эффекту от лечения, кроме того, введение экзогенного инсулина приводит к снижению секреции собственного инсулина. В исследовании А. Stride и соавт. не было выявлено разницы между уровнем HbA1c у пациентов, получающих сахароснижающую терапию (6,5%) и диетотерапию (6,4%). При отмене сахароснижающей терапии (инсулин и ПССП) через 3 мес уровень HbA1c не повышался [12]. Таким образом, пациентам с GCK-MODY не показана терапия инсулином или ПССП [1][32]. Если пациент получает сахароснижающую терапию при выявлении гетерозиготных мутаций в гене GCK, возможна отмена сахароснижающей терапии, в том числе инсулина [12]. В нашем исследовании верификация диагноза позволила снизить долю пациентов, получающих терапию, с 15,4 до 7,7%.
У детей нарушения углеводного обмена при GCK-MODY диагностируются чаще всего случайно, асимптоматически в любом возрасте, в том числе с рождения, характеризуются сочетанием НГН и НТГ и, как правило, не требуют назначения сахароснижающей терапии. Основными догенетическими критериями диагностики MODY являются: возраст диагностики СД 10–45 лет, отягощенная наследственность по аутосомно-доминантному типу, отсутствие потребности или небольшая потребность в инсулине при длительности заболевания (3 года и более), отсутствие ожирения и ИР, отсутствие повышения АТ. Однако анализ собственных данных и литературы показал, что не всегда GCK-MODY соответствует всем критериям MODY. При мутациях de novo, асимптоматическом течении нарушений углеводного обмена или низкой осведомленности о состоянии здоровья родственников может отсутствовать положительный семейный анамнез по СД. Ожирение при GCK-MODY может встречаться с популяционной частотой. У детей с GCK-MODY отмечается пубертатная ИР, встречающаяся и у здоровых детей. Нарушения углеводного обмена при GCK носят неиммунный характер, однако при выявлении персистирующей гипергликемии натощак у детей с невысоким титром одного или двух типов специфических панкреатических АТ при отягощенной наследственности по СД необходимо также проводить исследование гена GCK. Данный подход к диагностике GCK-MODY приведет к более частому выявлению новых случаев заболевания.
Источники финансирования. Исследование выполнено в рамках государственного задания «Персонализированный подход к прогнозированию развития и дифференциальной диагностике сахарного диабета 1 типа у детей и подростков», регистрационный номер АААА-А20-120012190131-9.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Сечко Е.А. — концепция, получение, анализ данных, интерпретация результатов, написание статьи; Кураева Т.Л. — концепция, получение, анализ данных, интерпретация результатов, написание статьи; Зильберман Л.И. — концепция, внесение в рукопись существенной (важной) правки с целью повышения научной ценности статьи; Лаптев Д.Н. — концепция, внесение в рукопись существенной (важной) правки с целью повышения научной ценности статьи; Безлепкина О.Б. — концепция, внесение в рукопись существенной (важной) правки с целью повышения научной ценности статьи; Петеркова В.А. — концепция, внесение в рукопись существенной (важной) правки с целью повышения научной ценности статьи. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
1. Hattersley AT, Greeley SAW, Polak M, et al. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19(S27):47-63. doi: https://doi.org/10.1111/pedi.12772
2. Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24(1):44-53. doi: https://doi.org/10.2337/diab.24.1.44
3. Yamagata K, Furuta H, Oda N, et al. Mutations in the hepatocyte nuclear factor-4α gene in maturity-onset diabetes of the young (MODY1). Nature. 1996;384:458-460. doi: https://doi.org/10.1038/384458a0
4. Mozzillo E, Salzano G, Barbetti F, et al. Survey on etiological diagnosis of diabetes in 1244 Italian diabetic children and adolescents: impact of access to genetic testing. Diabetes Res Clin Pract. 2015;107(3):e15-e18. doi: https://doi.org/10.1016/j.diabres.2015.01.003
5. Fendler W, Borowiec M, Baranowska-Jazwiecka A, et al. Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia. 2012;55(10):2631-2635. doi: https://doi.org/10.1007/s00125-012-2621-2
6. Chakera AJ, Spyer G, Vincent N, et al. The 0.1% of the population with glucokinase monogenic diabetes can be recognized by clinical characteristics in pregnancy: the Atlantic Diabetes in Pregnancy cohort. Diabetes Care. 2014;37(5):1230-1236. doi: https://doi.org/10.2337/dc13-2248
7. Кураева Т.Л., Сечко Е.А., Зильберман Л.И., и др. Молекулярно-генетические и клинические варианты MODY2 и MODY3 у детей в России // Проблемы эндокринологии. — 2015. — Т. 61. — №5. — С. 14-25. doi: https://doi.org/10.14341/probl201561514-25
8. Зубкова Н.А., Гиоева О.А., Тихонович Ю.В., и др. Клиническая и молекулярно-генетическая характеристика случаев MODY1-3 в Российской Федерации, выявленных по результатам NGS // Проблемы эндокринологии. — 2017. — Т. 63. — №6. — С. 369-378. doi: https://doi.org/10.14341/probl2017636369-378
9. Glotov OS, Serebryakova EA, Turkunova ME, et al. Whole-exome sequencing in Russian children with non-type 1 diabetes mellitus reveals a wide spectrum of genetic variants in MODY-related and unrelated genes. Mol Med Rep. 2019;20(6):4905-4914. doi: https://doi.org/10.3892/mmr.2019.10751
10. Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes. 2002;51(S3):S394-S404. doi: https://doi.org/10.2337/diabetes.51.2007.s394
11. Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45(3):427-435. doi: https://doi.org/10.1007/s00125-001-0770-9
12. Stride A, Shields B, Gill-Carey O, et al. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia. 2014;57(1):54-56. doi: https://doi.org/10.1007/s00125-013-3075-x
13. Feigerlová E, Pruhová S, Dittertová L, et al. Aetiological heterogeneity of asymptomatic hyperglycaemia in children and adolescents. Eur J Pediatr. 2006;165(7):446-452. doi: https://doi.org/10.1007/s00431-006-0106-3
14. Pihoker C, Gilliam LK, Ellard S, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab. 2013;98(10):4055-4062. doi: https://doi.org/10.1210/jc.2013-1279
15. Гиоева О.А., Колодкина А.А., Васильев Е.В., и др. Наследственный вариант сахарного диабета, обусловленного дефектом гена NEUROD1 (MODY6): первое описание в России // Проблемы Эндокринологии. — 2016. — Т. 62. — №3. — C. 16-20. doi: https://doi.org/10.14341/probl201662316-20
16. Osbak KK, Colclough K, Saint-Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30(11):1512-1526. doi: https://doi.org/10.1002/humu.21110
17. Hager J, Blanché H, Sun F, et al. Six mutations in the glucokinase gene identified in MODY by using a nonradioactive sensitive screening technique. Diabetes. 1994;43(5):730-733. doi: https://doi.org/10.2337/diab.43.5.730
18. Gragnoli C, Cockburn BN, Chiaramonte F, et al. Early-onset Type II diabetes mellitus in Italian families due to mutations in the genes encoding hepatic nuclear factor 1 alpha and glucokinase. Diabetologia. 2001;44(10):1326-1329. doi: https://doi.org/10.1007/s001250100644
19. Lehto M, Wipemo C, Ivarsson SA, et al. High frequency of mutations in MODY and mitochondrial genes in Scandinavian patients with familial early-onset diabetes. Diabetologia. 1999;42(9):1131-1137. doi: https://doi.org/10.1007/s001250051281
20. Lorini R, Klersy C, d’Annunzio G, et al. Maturity-onset diabetes of the young in children with incidental hyperglycemia: a multicenter Italian study of 172 families. Diabetes Care. 2009;32(10):1864-1866. doi: https://doi.org/10.2337/dc08-2018
21. Takeda J, Gidh-Jain M, Xu LZ, et al. Structure/function studies of human beta-cell glucokinase. Enzymatic properties of a sequence polymorphism, mutations associated with diabetes, and other site-directed mutants. J Biol Chem. 1993;268(20):15200-15204.
22. Ellard S, Beards F, Allen LI, et al. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia. 2000;43(2):250-253. doi: https://doi.org/10.1007/s001250050038
23. Bertini C, Maioli M, Fresu P, et al. A new missense mutation in the glucokinase gene in an Italian Mody family. Diabetologia. 1996;39(11):1413-1414.
24. Gidh-Jain M, Takeda J, Xu LZ, et al. Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proc Natl Acad Sci U S A. 1993;90(5):1932-1936. doi: https://doi.org/10.1073/pnas.90.5.1932
25. Mantovani V, Salardi S, Cerreta V, et al. Identification of eight novel glucokinase mutations in Italian children with maturity-onset diabetes of the young. Hum Mutat. 2003;22(4):338. doi: https://doi.org/10.1002/humu.9179
26. Velho G, Blanché H, Vaxillaire M, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia. 1997;40(2):217-224. doi: https://doi.org/10.1007/s001250050666
27. Galán M, Vincent O, Roncero I, et al. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem J. 2006;393(Pt1):389-396. doi: https://doi.org/10.1042/BJ20051137
28. McKinney JL, Cao H, Robinson JF, et al. Spectrum of HNF1A and GCK mutations in Canadian families with maturity-onset diabetes of the young (MODY). Clin Invest Med. 2004;27(3):135-141.
29. Barrio R, Bellanné-Chantelot C, Moreno JC, et al. Nine novel mutations in maturity-onset diabetes of the young (MODY) candidate genes in 22 Spanish families. J Clin Endocrinol Metab. 2002;87(6):2532-2539. doi: https://doi.org/10.1210/jcem.87.6.8530
30. Jetton TL, Liang Y, Pettepher CC, et al. Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J Biol Chem. 1994;269(5):3641-3654. doi: https://doi.org/10.1016/S0021-9258(17)41910-7
31. Njølstad PR, Søvik O, Cuesta-Muñoz A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344(21):1588-1592. doi: https://doi.org/10.1056/NEJM200105243442104
32. Velho G, Petersen KF, Perseghin G, et al. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J Clin Invest. 1996;98(8):1755-1761. doi: https://doi.org/10.1172/JCI118974
33. Овсянникова А.К., Шахтшнейдер Е.В., Иванощук Д.Е., и др. Течение сахарного диабета взрослого типа у молодых лиц старше 18 лет, обусловленного мутацией гена глюкокиназы (GCK-MODY): данные проспективного наблюдения // Сахарный диабет. — 2021. — Т. 24. — №2. — С. 133-140. doi: https://doi.org/10.14341/DM12319
34. Hughes AE, De Franco E, Globa E, et al. Identification of GCK-maturity-onset diabetes of the young in cases of neonatal hyperglycemia: A case series and review of clinical features. Pediatr Diabetes. 2021;22(6):876-881. doi: https://doi.org/10.1111/pedi.13239
35. Дедов И.И., Зубкова Н.А., Арбатская Н.Ю., и др. MODY2: клинические и молекулярно-генетические характеристики 13 случаев заболевания. Первое описание МОDY в России // Проблемы Эндокринологии. — 2009. — Т. 55. — №3. — C. 3-7. doi: https://doi.org/10.14341/probl20095533-7
36. Steele AM, Wensley KJ, Ellard S, et al. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS One. 2013;8(6):e65326. doi: https://doi.org/10.1371/journal.pone.0065326
37. Тутельян В.А., Батурин А.К., Конь И.Я., и др. Распространенность ожирения и избыточной массы тела среди детского населения РФ: мультицентровое исследование // Педиатрия. Журнал им. Г.Н. Сперанского. — 2014. — Т. 93. — №5. — С. 28-31.
38. Kleinberger JW, Copeland KC, Gandica RG, et al. Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes: the TODAY clinical trial. Genet Med. 2018;20(6):583-590. doi: https://doi.org/10.1038/gim.2017.150
39. Lehto M, Tuomi T, Mahtani MM, et al. Characterization of the MODY3 phenotype. Early-onset diabetes caused by an insulin secretion defect. J Clin Invest. 1997;99(4):582-591. doi: https://doi.org/10.1172/JCI119199
40. Wilkin TJ. The accelerator hypothesis: weight gain as the missing link between Type I and Type II diabetes. Diabetologia. 2001;44(7):914-922. doi: https://doi.org/10.1007/s001250100548
41. Clément K, Pueyo ME, Vaxillaire M, et al. Assessment of insulin sensitivity in glucokinase-deficient subjects. Diabetologia. 1996;39(1):82-90. doi: https://doi.org/10.1007/BF00400417
42. Gungor N, Saad R, Janosky J, Arslanian S. Validation of surrogate estimates of insulin sensitivity and insulin secretion in children and adolescents. J Pediatr. 2004;144(1):47-55. doi: https://doi.org/10.1016/j.jpeds.2003.09.045
43. Stanik J, Dusatkova P, Cinek O, et al. De novo mutations of GCK, HNF1A and HNF4A may be more frequent in MODY than previously assumed. Diabetologia. 2014;57(3):480-484. doi: https://doi.org/10.1007/s00125-013-3119-2
44. Spyer G, Macleod KM, Shepherd M, et al. Pregnancy outcome in patients with raised blood glucose due to a heterozygous glucokinase gene mutation. Diabet Med. 2009;26(1):14-18. doi: https://doi.org/10.1111/j.1464-5491.2008.02622.x
Сечко Елена Александровна, кандидат медицинских наук
117036, Москва, ул. Дм. Ульянова, д. 11
eLibrary SPIN: 4608-565
Кураева Тамара Леонидовна, доктор медицинских наук, проф.
Москва
eLibrary SPIN: 8206-0406
Зильберман Любовь Иосифовна, кандидат медицинских наук
Москва
eLibrary SPIN: 4488-7724
Лаптев Дмитрий Никитич, доктор медицинских наук
Москва
eLibrary SPIN: 2419-4019
Безлепкина Ольга Борисовна, доктор медицинских наук, профессор
Москва
eLibrary SPIN: 3884-0945
Петеркова Валентина Александровна, доктор медицинских наук, профессор, академик РАН
Москва
eLibrary SPIN: 4009-2463
|
|
1. Рисунок 1. Корреляционная взаимосвязь между возрастом обследования и уровнем гликемии натощак у детей с GCK-MODY. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(131KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Корреляционная взаимосвязь между возрастом обследования и уровнем С-пептида натощак у детей с GCK-MODY. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(109KB)
|
Метаданные ▾ | |
|
|
3. Рисунок 3. Корреляционная взаимосвязь между возрастом обследования и уровнем инсулина натощак у детей с GCK-MODY. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(140KB)
|
Метаданные ▾ | |
Сечко Е.А., Кураева Т.Л., Зильберман Л.И., Лаптев Д.Н., Безлепкина О.Б., Петеркова В.А. Неиммунный сахарный диабет у детей, обусловленный гетерозиготными мутациями в гене глюкокиназы (GCK-MODY): анализ данных 144 пациентов. Сахарный диабет. 2022;25(2):145-154. https://doi.org/10.14341/DM12819
Sechko E.A., Kuraeva T.L., Zilberman L.I., Laptev D.N., Bezlepkina O.B., Peterkova V.A. Non-immune diabetes mellitus in children due to heterozygous mutations in the glucokinase gene (GCK-MODY): data of 144 patients. Diabetes mellitus. 2022;25(2):145-154. (In Russ.) https://doi.org/10.14341/DM12819

 |
Адрес: 117036, Российская Федерация, Москва, улица Дмитрия Ульянова, дом 11.
Обработка персональных данных







































