




Содержание
Перейти к:
https://doi.org/10.14341/DM13314
Перейти к:
Недостаточная осведомленность врачебного сообщества о наследственных липодистрофиях затрудняет выявляемость данной патологии. Несмотря на прогресс в понимании молекулярно-генетической основы различных синдромов липодистрофии, многие пациенты по-прежнему ускользают от внимания врачей и узнают о своем заболевании уже на поздних стадиях. Американская ассоциация клинической эндокринологии (American Association of Clinical Endocrinology, AACE) создала экспертную рабочую группу, в которую вошли практикующие врачи и лидеры в области лечения и исследования синдромов липодистрофии, для разработки клинических рекомендаций.
Синдромы липодистрофии представляют собой гетерогенную группу чрезвычайно редких заболеваний, общим для которых является дефицит жировой ткани при отсутствии алиментарной депривации или катаболического состояния. Наследственные липодистрофии связанны с потенциально серьезными метаболическими осложнениями, включающими сахарный диабет, гипертриглицеридемию, стеатоз печени, синдром поликистозных яичников и черный акантоз. Синдромы липодистрофии неоднородны и диагностируются на основании совокупности фенотипических проявлений, данных клинического обследования и дополняются результатами генетического тестирования. Пациентам с установленным диагнозом необходимо проводить ежегодный скрининиг на наличие ассоциированных с данными синдромами заболеваний с целью более раннего их выявления. По данным литературы, распространенность синдромов наследственных липодистрофий оценивается как 1 случай на 1 млн населения.
Ввиду их редкой встречаемости большинство клиницистов не знакомы с данным диагнозом и принципами его лечения. В связи с чем повышение осведомленности врачей различных специальностей о такой орфанной патологии имеет крайне важное значение для более ранней диагностики, что определило цели данной публикации.
Фролкова Н.В., Кокшарова Е.О., Мишина Е.Е., Шестакова М.В. Наследственные липодистрофии: как не пропустить диагноз? Сахарный диабет. 2025;28(4):413-423. https://doi.org/10.14341/DM13314
Frolkova N.V., Koksharova E.O., Mishina E.E., Shestakova M.V. Hereditary lipodystrophies: how not to miss the diagnosis? Diabetes mellitus. 2025;28(4):413-423. (In Russ.) https://doi.org/10.14341/DM13314
Синдромы липодистрофии представляют собой гетерогенную группу редких заболеваний, характеризующихся дефицитом подкожно-жировой клетчатки (ПЖК), что приводит к эктопическому накоплению липидов в печени, мышцах и других органах, вызывая резистентность тканей к инсулину и приводя к потенциально опасными для жизни состояниям. Развивающиеся метаболические и гормональные нарушения ассоциированы с развитием сахарного диабета (СД), гипертриглицеридемии (ГТГ), метаболически ассоциированной жировой болезни печени (МАЖБП) и синдрома поликистозных яичников (СПКЯ). В связи с чем уровень смертности среди пациентов этой группы достаточно высок [1][2].
По оценкам различных исследователей, распространенность синдромов липодистрофии составляет от 1,3 до 4,7 случая на 1 млн в мире [2]. Однако в США был проведен обзор медицинских карт, по результатам которого клиническая распространенность липодистрофий была оценена как 1 на 20 000 человек, а генетическая — 1 на 7000 человек, что указывает на то, что распространенность липодистрофий может быть значительно выше, чем предполагалось [3].
Липодистрофии можно классифицировать по этиологии (наследственные или приобретенные) и степени потери ПЖК (генерализованная (ГЛ) или парциальная (ПЛ)). На основании этого выделяют 4 варианта липодистрофий (табл. 1) [1][4–6].
Таблица 1. Классификация липодистрофий
|
Тип |
Мутантный ген и кодируемый белок |
Механизм наследования |
|
Врожденные генерализованные липодистрофии (Синдром Берардинелли-Сейпа) |
||
|
Тип 1 |
AGPAT2, фермент 1-ацилглицерол-3-фосфат-О-ацилтрансфераза 2 |
Аутосомно-рецессивный |
|
Тип 2 |
BSCL2, белок сейпин |
Аутосомно-рецессивный |
|
Тип 3 |
CAV1, белок кавеолин 1 типа |
Аутосомно-рецессивный |
|
Тип 4 |
PTRF, белок кавин |
Аутосомно-рецессивный |
|
Семейные парциальные липодистрофии (Синдром Даннигана-Кобберлинга) |
||
|
Тип 1 (синдром Кобберлинга) |
Неизвестен |
Неизвестен |
|
Тип 2 (синдром Даннигана) |
LMNA, лапин А и С |
Аутосомно-доминантный |
|
Тип 3 (синдром Даннигана) |
PPARG, PPARγ |
Аутосомно-доминантный |
|
Тип 4 |
PLIN1, перилипин 1 |
Аутосомно-доминантный |
|
Тип 5 |
CIDEC, эффектор С, подобный фактору фрагментации ДНК, индуцирующему клеточную гибель |
Аутосомно-рецессивный |
|
Тип 6 |
LIPE, гормончувствительная липаза |
Аутосомно-рецессивный |
|
Тип 7 |
AKT2, протеинкиназа В |
Аутосомно-доминантный |
|
Многокомпонентные генетические синдромы, сопровождающиеся липодистрофией |
||
|
Истинная прогерия Хатчинсона-Гилфорда |
LMNA, ламин А |
Аутосомно-рецессивный |
|
Синдром Вернера (прогерия взрослых) |
WRN, ДНК-хеликаза |
Аутосомно-рецессивный |
|
Атипичная прогерия, атипичный синдром Вернера |
LMNA, ламин А |
Аутосомно-доминантный |
|
Мандибулоакральная дисплазия (МАД): - тип А - тип B |
LMNA ZMPSTE24 |
Аутосомно-рецессивный |
|
Мандибулярная гипоплазия, глухота и ПС |
POLD1, каталитическая субъединица гена полимеразы дельта 1 |
Аутосомно-доминантный |
|
Прогероидный cиндром |
SPRTN, белок SprT-подобного N-терминального домена |
Аутосомно-рецессивный |
|
Синдром Нестера-Гильермо |
BANF1, фактор-1-барьера аутоинтеграции |
Аутосомно-рецессивный |
|
Синдром Марфана с ПС и липодистофией |
FBN1, фибриллин 1 |
Аутосомно-доминантный |
|
Синдром Keppen-Lubinsky |
KCNJ6 |
Аутосомно-доминантный |
|
SHORT-синдром |
PIK3R1 |
Аутосомно-доминантный |
|
CANDLE-синдром |
PSMB8 |
Аутосомно-рецессивный |
|
Приобретенные генерализованные липодистрофии (Синдром Лоуренса-Сейпа) • Панникулит-ассоциированные • Аутоиммунные • Идиопатические |
||
|
Приобретенные парциальные липодистрофии (Синдром Барракера-Симонса) • Ассоциированный с мембранопролиферативным гломерулонефритом • Аутоиммунные • Идиопатические |
||
|
Липодистрофия у ВИЧ-инфицированных пациентов, индуцированная высокоактивной антиретровирусной терапией |
||
Помимо этих основных вариантов, некоторые другие редкие синдромы также могут проявляться липодистрофией. Различные прогероидные синдромы могут иметь аутосомно-рецессивное (LMNA, ZMPSTE24, SPRTN, WRN, BANF1) или доминантное наследование (LMNA, FBN1, CAV1, POLD1, KCNJ6) и проявляться парциальными или генерализованными липодистрофиями наряду с прогероидными особенностями фенотипа и различными метаболическими осложнениями [4].
Потеря ПЖК может варьироваться от генерализованной (затрагивающей почти все жировые отложения тела) до парциальной (затрагивающей только конечности или верхнюю часть тела) или локализованной на небольших участках (рис. 1) [1][4]. Тяжесть метаболических осложнений обычно связана со степенью потери жира в организме [4].
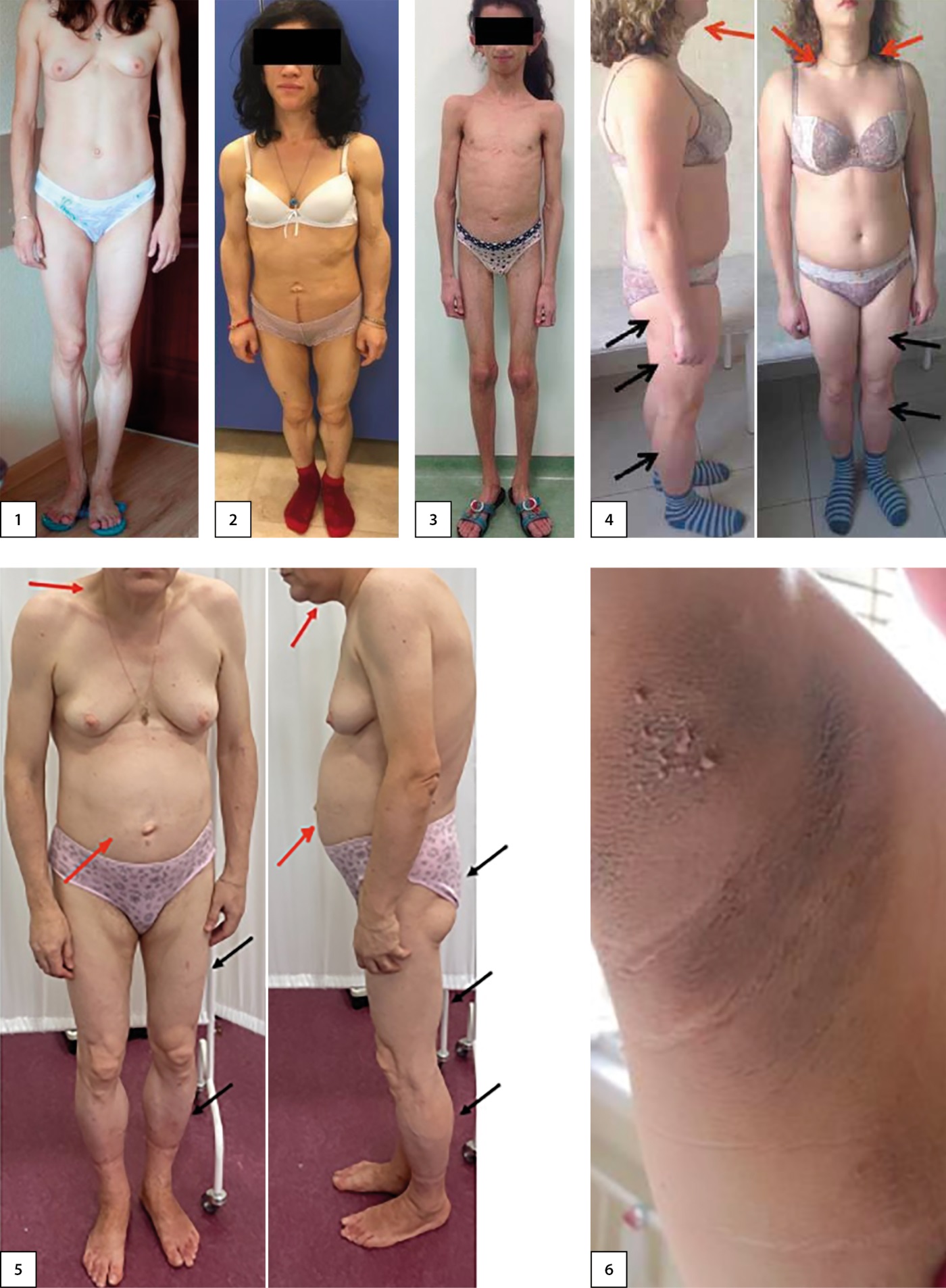
Рисунок 1. Фенотип пациенток с различными формами наследственных липодистрофий (из архива ГНЦ РФ ФГБУ «НМИЦ эндокринологии им. академика И.И. Дедова» Минздрава России). На 1 и 2 фотографиях — пациентки с генерализованными липодистрофиями, фотография 3 — атипичный прогероидный синдром (мутация в гене LMNA), фотографии 4 и 5 — пациентки с семейными парциальными липодистрофиями, фотография 6 — acanthosis nigricans подмышечной впадины. Красной стрелкой обозначены зоны липогипертрофии; черной — зоны липодистрофии.
В данном обзоре мы хотим обратить внимание клиницистов на распространенность различных метаболических нарушений и патологий со стороны различных органов и систем, характерных для наследственных липодистрофий: врожденной генерализованнной липодистрофии и семейной парциальной липодистрофии.
Врожденная генерализованная липодистрофия, или синдром Берардинелли-Сейпа была впервые описана Berardinelli W. и соавт. в 1954 г. у двух братьев из Бразилии в возрасте 2 и 6 лет, у которых наблюдались выраженная гепатоспленомегалия, акромегалоидный гигантизм, жировая дистрофия печени и гипертриглицеридемия (ГТГ) [7][8]. Впоследствии в Норвегии в 1959 г. Seip M. и соавт. выявили еще трех пациентов, у которых с рождения наблюдался генерализованный дефицит жира в организме [7][8]. ВГЛ — это очень редкое заболевание, с момента первых сообщений в 50-х годах по настоящее время в литературе представлено описание около 300–500 пациентов [8]. Точная распространенность ВГЛ в популяции неизвестна, большинство исследователей оценивают ее как 1 на 10 млн человек в мире, 1 на 12 млн в США, 1 на 1 млн — в Норвегии, 1 на 500 тыс. — в Португалии и 1 на 200 тыс. — в Ливане [9]. На сегодняшний день данные о распространенности врожденной генерализованной липодистрофии в России отсутствуют.
При ВГЛ характерно избыточное накопление триглицеридов (ТГ) в печени и скелетных мышцах в связи с невозможностью их депонирования в жировой ткани, это является основной причиной метаболических осложнений при данном синдроме. Крайне низкие уровни лептина еще больше усугубляют метаболические нарушения и вызывают неконтролируемый аппетит у пациентов [8].
В 1974 г. Dunnigan MG. описал клинический случай СПЛ, сопровождающейся метаболическими нарушениями. Спустя год Köbberling представил другой вариант ПЛ, при котором дефицит ПЖК в основном затрагивал верхние и нижние конечности [10].
Клинический фенотип СПЛ обычно развивается в период полового созревания с заметной потерей подкожного жира в области конечностей, туловища и ягодиц и избыточным его накоплением на лице, шее, спине, животе и в области больших половых губ [11].
Диагностика липодистрофии проводится преимущественно путем сбора анамнеза и осмотра пациента, и может быть затруднена, особенно при парциальных формах, так как клинический фенотип больных крайне гетерогенен, а особенности внешности могут быть объяснены индивидуальными особенностями, спортивным телосложением и т.д. Постановка диагноза особенно трудна у мужчин, которые обычно имеют худощавое телосложение с выраженным рельефом мускулатуры, и у детей препубертатного возраста из-за отсутствия явных клинических признаков [12].
Большинство исследователей ранее оценивали распространенность наследственных липодистрофий от 1,3 до 4,78 на 1 млн населения, однако недавно она была повторно оценена как минимум 1 на 20 тыс. человек, что позволяет убедиться в значительном числе недиагностированных случаев липодистрофии [13].
По данным исследования [6] Y. Handelsman и соавт., в котором приняли участие более 5000 голландских пациентов с СД, из 24 пациентов, соответствующих критериям включения (индекс массы тела (ИМТ)<27 кг/м² и использование >100 единиц инсулина в день), у 5 пациентов в конечном итоге была диагностирована СПЛ (у трех — с подтвержденными генетическими мутациями). Таким образом, при более полном понимании спектра проявлений липодистрофии и связи между генными мутациями и клиническими проявлениями общепринятая диагностическая концепция может потребовать модификации и расширения, особенно в отношении форм парциальной липодистрофии.
В исследовании [13] Mosbah H. и соавт. приняли участие 109 пациентов с синдромами липодистрофий, из которых СПЛ имели 93 пациента (подавляющее большинство — женщины — 86%), а ВГЛ — 16 (75% женщин). У большинства включенных в исследование пациентов регистрировались сопутствующие метаболические заболевания, такие как: СД, ГТГ, стеатоз печени, нарушения со стороны сердечно-сосудистой системы и гинекологические осложнения. Средний возраст на момент постановки диагноза составил 30 [ 23; 47] лет. В целом диагноз липодистрофии считали «запоздалым» у 71% пациентов (59 человек из 83 опрошенных), а диагностический процесс ими воспринимался как «очень трудный». Учитывая всех респондентов, медиана задержки постановки диагноза составила 12 [ 5; 25] лет, при этом 36% респондентов консультировались более чем у пяти разных врачей перед постановкой диагноза по поводу симптомов, связанных с липодистрофией. Количество врачей, посещенных до постановки диагноза, не различалось в зависимости от типа липодистрофии, пола, уровня образования и профессиональной деятельности пациентов. Перед окончательным диагнозом 91% пациентов обращался к своему семейному врачу по поводу симптомов, связанных с липодистрофией, 72% — к эндокринологу, 60% — к кардиологу, 59% — к дерматологу и 54% — к диетологу. Перед постановкой диагноза по поводу симптомов, связанных с липодистрофией, 69% женщин консультировались с гинекологом. Эндокринолог поставил диагноз 77% больных.
При проведении дифференциальной диагностики должны учитываться состояния, которые сопровождаются значительной потерей массы тела, такие как недоедание, декомпенсация сахарного диабета, тиреотоксикоз, надпочечниковая недостаточность, нервная анорексия, раковая кахексия, ВИЧ-ассоциированное истощение и хронические инфекции [5].
Генерализованные липодистрофии могут быть ошибочно приняты за мутации рецептора инсулина или акромегалию/гигантизм, в то время как семейные парциальные липодистрофии могут быть ошибочно диагностированы как синдром Кушинга, алиментарное ожирение или множественный симметричный липоматоз [5].
Дифференциальная диагностика наследственных липодистрофий проводится со следующими эндокринными заболеваниями.
Как при наследственных липодистрофиях, так и при акромегалии, в организме пациентов анаболические процессы преобладают над катаболическими. Вследствие этого у данных групп пациентов отмечаются схожие клинические проявления: расширение межзубных промежутков, висцеро- и флебомегалия, умеренный прогнатизм, укрупнение кистей и стоп, затылочная кожная складка, утолщение всех слоев дермы нарушение углеводного и липидного обменов. При этом причины избыточных анаболических процессов различны. При акромегалии данные процессы обусловлены гиперпродукцией гормона роста, а у больных наследственными липодистрофиями — хронической эндогенной гиперпродукцией инсулина [14].
Матронизм, повышение артериального давления (АД), нарушение толерантности к глюкозе (НТГ), признаки вирилизации — общие проявления эндогенного гиперкортицизма и наследственных липодистрофий. Матронизм при наследственных липодистрофиях объясняется тем, что у пациентов сохраняется способность депонировать нейтральные жиры в адипоцитах, расположенных исключительно в области лица, шеи и надключичных впадин. Резистентность к гипогликемическому действию инсулина связана с ГТГ и приводит к нарушению углеводного обмена. Вирилизация при наследственных липодистрофиях имеет яичниковое происхождение, в то время как у больных эндогенным гиперкортицизмом это связано с избыточным синтезом андрогенов надпочечниками. При проведении дифференциальной диагностики необходимо обратить внимание на следующие моменты. Для наследственных липодистрофий характерна мышечная гипертрофия, особенно конечностей. Фенотипу пациентов с эндогенным гиперкортицизмом, напротив, свойственны уменьшение мышечной массы и гипотрофия мышц конечностей. При эндогенном гиперкортицизме кожные покровы истончены вследствие белковой дистрофии, характерны стрии, усиление сосудистого рисунка, гнойничковые поражения волосяных фолликулов [4][14].
У больных СД в состоянии тяжелой декомпенсации может почти полностью исчезнуть подкожный жировой слой, что и является причиной диагностических ошибок. К общим признакам для обоих заболеваний следует отнести и значительную потребность в экзогенном инсулине. В случае наследственных липодистрофий необходимость введения очень больших доз инсулина постоянна и связана с гиперлипидемией и значительной периферической инсулинорезистентностью. Во втором случае суточная доза вводимого инсулина существенно возрастает только на период декомпенсации углеводного и жирового обменов. Основным дифференциально-диагностическим признаком является отсутствие кетоацидоза у пациентов с наследственными липодистрофиями, для них также не характерны полиурия и полидипсия, столь типичные для декомпенсации углеводного обмена при СД [14].
Патогенез экзогенно-конституционального ожирения включает несколько взаимосвязанных процессов, которые способствуют развитию метаболических нарушений. Избыточное потребление высококалорийной пищи приводит к накоплению висцерального жира, что способствует образованию свободных жирных кислот и провоспалительных молекул. Избыточная секреция провоспалительных цитокинов, таких как TNF-α, интерлейкин-6, а также гормонов, например, резистина, приводит к хроническому воспалению в организме. Это, в свою очередь, приводит к инсулинорезистентности, резистентности к лептину, гипергликемии, дислипидемии и артериальной гипертензии (АГ) [15]. При синдромах липодистрофии потеря ПЖК и увеличение массы висцерального жира демонстрируют тот же патофизиологический механизм развития инсулинорезистентности одновременно с нарушением метаболизма ТГ. Несмотря на различие в уровнях лептина и адипонектина при висцеральном ожирении (центральная резистентность к лептину) и липодистрофиях (нехватка синтеза из-за снижения массы адипоцитов), оба состояния сопровождаются выраженной инсулинорезистентностью, которая возникает вторично вследствие избыточной секреции адипоцитов и эктопического накопления жира в печени, скелетных мышцах и других тканях — мишенях инсулина (табл. 2) [16].
Таблица 2. Сравнение метаболического синдрома при экзогенно-конституциональном ожирении и синдромах липодистрофии
|
Экзогенно-конституциональное ожирение |
Синдромы липодистрофии |
|
Выраженное увеличение жировой массы |
Значительное снижение жировой ткани |
|
Чем больше избыточная жировая масса, тем более выражен фенотип метаболического синдрома |
Чем больше снижение жировой массы, тем более выражен фенотип метаболического синдрома |
|
Инсулинорезистентность/СД |
|
|
Метаболический синдром (нарушение углеводного обмена, ГТГ, низкий уровень ЛПВП, стеатоз печени, acanthosis nigricans, СПКЯ, абдоминальное ожирение, АГ) |
|
|
Липотоксичность |
|
Примечание. АГ — артериальная гипертензия; ЛПВП — липопротеиды высокой плотности; СД — сахарный диабет; СПКЯ — синдром поликистозных яичников; СД — сахарный диабет, ГТГ — гипертриглицеридемия.
Одним из наиболее распространенных метаболических нарушений при наследственных липодистрофиях является нарушение углеводного обмена, которое может варьировать от предиабета до декомпенсированного СД вследствие выраженной инсулинорезистентности. Из-за снижения объема жирового депо развивается ГТГ, происходит эктопическое отложение липидов в клетках печени (МАЖБП) и мышечной ткани, что ведет к инсулинорезистентности и компенсаторной гиперинсулинемии.
Ajluni N. и соавт. провели исследование [17], в которое были включены 23 пациента (22 с СПЛ, 1 с ППЛ, 78,3% из них — женщины в возрасте от 12 до 64 лет). У всех включенных пациентов был либо СД (82,6%), либо предиабет (17,4%) с различной степенью контроля. Среднее значение гликированного гемоглобина (HbA1c) составило 8,6±1,9%. 19 пациентов (82,6%) использовали сахароснижающие препараты. 10 пациентов (43,5%) использовали комбинированную терапию с двумя или более средствами. Инсулинотерапию использовали 12 пациентов (52,2%), и 9 из этих пациентов требовалось более 100 единиц в день. Шесть пациентов (26,1%) использовали высококонцентрированный инсулин U-500.
Akinci B. и соавт. [2] обследовали 230 пациентов с ГЛ и ПЛ, у 58,3% из них был выявлен СД или инсулинорезистентность. Авторы отметили, что у многих пациентов в их выборке СД был диагностирован значительно раньше, чем им был поставлен диагноз «ГЛ» или «ПЛ», что также указывает на необходимость повышения осведомленности о синдромах липодистрофии среди медицинского сообщества.
Дислипидемия у пациентов с наследственными липодистрофиями характеризуется ГТГ и низким уровнем липопротеинов высокой плотности (ЛПВП). У некоторых пациентов отмечаются крайне высокие значения ТГ и хиломикронемия, что приводит к рецидивирующим приступам острого панкреатита, липемии сетчатки, а также образованию туберозных и эруптивных ксантом. У некоторых пациентов наблюдались плоские ксантомы на ладонях и подошвенных поверхностях стоп [1][18]. ГТГ обычно имеет легкую или умеренную степень тяжести при СПЛ и, вероятно, играет роль в раннем развитии атеросклеротических сердечно-сосудистых заболеваний (АСССЗ) [11].
ГТГ присутствует более, чем у 70% пациентов с ВГЛ и развивается в основном в позднем детском и подростковом возрасте и только в редких случаях в младенчестве [8].
Lazarte J. и соавт. определили распространенность тяжелой ГТГ и панкреатита среди группы из 74 пациентов с СПЛ [11]. Авторы обнаружили, что общая распространенность тяжелой ГТГ и госпитализации по поводу острого панкреатита составила 5,4 и 4,1% соответственно. Среди пациентов с сопутствующим СД и без него медиана уровня ТГ составила 2,73 и 1,86 ммоль/л соответственно. Среди пациентов с СПЛ и СД распространенность тяжелой ГТГ (>10 ммоль/л) и панкреатита составила 14,3% и 10,7% соответственно, по сравнению с отсутствием случаев любого из осложнений у пациентов с СПЛ без диабета. Это указывает на то, что риск тяжелой ГТГ и панкреатита при СПЛ зависит от сочетанного наличия диабета [11]. Ранее исследователи провели оценку распространенности панкреатита среди пациентов с СПЛ, которая составила 11,9%. В исследовании приняли участие 149 пациентов с СПЛ, из которых у 68 был СПЛ 2 типа [11].
В указанном ранее исследовании Ajluni N. и соавт. в общей сложности у 22 из 23 пациентов была выявлена ГТГ, средний показатель 11,9±19,7 ммоль/л. У шести пациентов были очень высокие показатели ТГ — более 11,3 ммоль/л. Большинство (17 пациентов) принимали гиполипидемические препараты. Рецидивирующий панкреатит наблюдался у 7 из 23 пациентов (30,4%). У трех пациентов эпизод острого панкреатита наблюдался при уровне ТГ менее 9 ммоль/л [17].
Спектр заболеваний начинается с бессимптомного накопления жира в гепатоцитах (стеатоз) и может прогрессировать до стеатогепатита с фиброзом или без и даже цирроза и гепатоцеллюлярного рака [18][19]. Метаболически ассоциированная жировая болезнь печени (МАЖБП) — часто встречающееся заболевание печени у пациентов с наследственными липодистрофиями. 82% пациентов, учувствовавших в исследовании Safar Zadeh E. [19] имели МАЖБП. Авторы оценивали степень фиброза печени по результатам биопсии, более тяжелая степень отмечалась у пациентов с ВГЛ 2 типа.
Lüdtke A. и соавт. провели исследование [20] с целью определения распространенности стеатоза печени у пациентов с СПЛ. Авторы обследовали 18 пациентов из шести семей с СПЛ 2 типа — у всех была диагностирована МАЖБП (стеатоз) по данным УЗИ печени и подтверждена данными биопсии печени у 2 пациентов. Среди участников этого исследования у 14 из 18 пациентов было выявлено нарушение углеводного обмена (предиабет/СД), а у 16 из 18 пациентов была обнаружена дислипидемия IIb, IV или V типа по Фредериксону. Предполагается, что эти метаболические нарушения (ГТГ и нарушение углеводного обмена), ассоциированные с инсулинорезистентностью, играют основную роль в многофакторном патогенезе стеатоза печени. Эктопическое отложение жира в тканях — мишенях инсулина, таких как печень, скелетные мышцы и бета-клетки, способствует тяжелой инсулинорезистентности, наблюдаемой при СПЛ [21].
Осведомленность врачей-кардиологов о синдромах наследственных липодистрофий имеет крайне важное значение для данной когорты пациентов. Сердечно-сосудистые заболевания значительно снижают качество жизни пациентов и могут приводить к летальному исходу в раннем возрасте. Своевременная постановка диагноза способствует более тщательному наблюдению, профилактике и выявлению патологии сердечно-сосудистой системы на более ранних этапах. В исследовании Mosbah H. и соавт. с участием 109 пациентов с наследственными липодистрофиями у 4 из 10 пациентов с ишемической болезнью сердца и у 6 из 13 пациентов с нарушениями сердечного ритма диагноз синдрома липодистрофии был установлен, соответственно, через 14 и 4 года после сердечно-сосудистого события [13].
В литературе имеется менее 20 публикаций об изменениях в работе сердца у пациентов с ВГЛ, размер выборки не превышает 10 пациентов. Эти исследования показали, что наиболее частыми сердечно-сосудистыми заболеваниями были дисфункция желудочков, гипертрофия миокарда, системная АГ, а также морфологические неспецифические фиброзные изменениями в миокарде [7, 22]. При гистологическом исследовании образцов также были выявлены патологические изменения коронарных артерий, в ряде случаев проявляющиеся инфарктом миокарда [22].
Гипертрофическая кардиомиопатия является хорошо известным проявлением синдрома ВГЛ и встречается у 20–25% пациентов. Средний возраст диагностики кардиомиопатии при синдроме ВГЛ составляет 20 лет. Механизм возникновения кардиомиопатии не ясен. Одним из возможных вариантов развития патологии является выраженная инсулинорезистентность. Предполагалось также прямое липотоксическое воздействие на кардиомиоциты, хотя отложение жира в миокарде четко не было продемонстрировано. Интересно, что кардиомиопатия чаще встречается при ВГЛ 2 типа, тогда как метаболические нарушения, в том числе гипергликемия, обычно более выражены у пациентов с ВГЛ 1 типа, что позволяет предположить вклад другого механизма, не связанного с гиперинсулинемией [23].
Среди проанализированных нами литературных данных при ВГЛ 1 типа исследователи сообщают о гипертрофической кардиомиопатии (4 из 21 пациента (19%) в исследовании Van Maldergem L. и соавт.) [24], гипертрофии левого желудочка (10 из 19 пациентов (53%) в исследовании Lupsa B. и соавт.) [25], ИБС (1 из 19 пациентов (5,3%) в исследовании Lupsa B. и соавт. [25] и 3 из 16 (18,37%) в исследовании Akinci B. и соавт. [26]); при ВГЛ 2 типа о гипертрофической кардиомиопатии (11 из 45 пациентов (24,4%) в исследовании Van Maldergem L. и соавт.) [24], АГ, кальцификации аортального клапана, гипертрофии левого желудочка (11 (50%), 2 (9,1%) и 12 (54,5%) из 22 пациентов соответственно в исследовании Guedes do Rêgo A. и соавт.) [7], смерти от сердечной недостаточности (3 из 45 пациентов (6,6%) Van Maldergem L. и соавт.) [24]. Пациенты с ВГЛ 4 типа предрасположены к серьезным аритмиям (10 из 30 зарегистрированных случаев ВГЛ 4 типа), таким как катехоламинергическая полиморфная желудочковая тахикардия (КПЖТ), длительный интервал QT, и были сообщения о внезапной смерти, вероятно, вторичной по отношению к желудочковым аритмиям [1][7].
Для пациентов с СПЛ нарушения со стороны сердечно-сосудистой системы наиболее подробно описаны для мутаций в гене LMNA (СПЛ 2 типа) как наиболее распространенного типа СПЛ. В исследовании Andre P. и соавт. [27] из 35 пациентов с СПЛ 2 типа у 10 (29%) были сердечно-сосудистые нарушения, среди которых: атрио-вентрикулярные блокады 1 и 2 степени (3 и 4 пациента соответственно), блокада левой ножки пучка Гиса (полная или неполная) — 6 пациентов, у 4 участников исследования был имплантируемый кардиовертер-дефибриллятор, желудочковые аритмии (1 случай неустойчивой желудочковой тахикардии и 1 — устойчивой желудочковой тахикардии). Hegele R.A. и соавт. описали у пациентов с СПЛ 2 типа значительно более частое возникновение ИБС (стабильная и нестабильная стенокардия, инфаркт миокарда, аортокоронарное шунтирование) по сравнению с контрольной группой (34,8 и 5,9% соответственно). Средний возраст постановки диагноза ИБС у пациентов с СПЛ 2 типа составил 46,5±3,8 года [28].
На важность тщательного кардиологического обследования пациентов с наследственными липодистрофиями указывает исследование, в котором у двух пациентов было зарегистрировано нарушение проводимости сердца, что привело к летальному исходу у одного из них, несмотря на предшествовавшую имплантацию кардиовертера-дефибриллятора [29].
Нередко с диагностикой наследственных липодистрофий могут столкнуться врачи-гинекологи при обследовании женщин, наблюдающихся по поводу СПКЯ или бесплодия. Считается, что гиперинсулинемия, вторичная по отношению к инсулинорезистентности, стимулирует тека-клетки к выработке андрогенов и, кроме того, снижает выработку глобулина, связывающего половые гормоны (ГСПГ) в печени, что приводит к повышению уровня свободного циркулирующего тестостерона [30][31].
У женщин с ВГЛ часто встречаются следующие клинические проявления: умеренный гирсутизм, клиторомегалия, нерегулярные менструации, а у некоторых наблюдается первичная или вторичная аменорея, снижение фертильности и СПКЯ [5][8][18][27][28]. Хотя опубликованные данные скудны, у детей с ГЛ могут наблюдаться раннее адренархе, истинное преждевременное половое созревание или центральный гипогонадизм [5][18]. Мужчины с ВГЛ могут иметь нормальную репродуктивную способность, однако у одного пациента зарегистрирован случай тератозооспермии [8].
Наиболее частой гинекологической патологией у женщин с липодистрофиями является СПКЯ, с предполагаемой распространенностью в литературе приблизительно 16–54% [32]. В многоцентровом проспективном наблюдательном исследовании, проведенном Akinci B. и соавт. [33] среди 24 пациентов с СПЛ ультразвуковые признаки поликистозных яичников имелись у 9 из 12 женщин репродуктивного возраста.
Другие гинекологические/акушерские осложнения, которые чаще встречаются у пациентов с СПЛ 2 типа, чем в общей популяции, включают бесплодие (28%), гестационный диабет (36%), выкидыши (50%), а также эклампсию и гибель плода (14%) [34].
Таким образом, роль гинеколога может быть очень ценной в диагностике синдромов липодистрофии у женщин.
Диабетическая нефропатия (ДН) представляет собой ведущую причину терминальной стадии хронической болезни почек (ХБП) в общей популяции. Поскольку интенсивность и продолжительность гипергликемии являются основными известными патогенетическими факторами ДН, можно было бы ожидать, что у пациентов с синдромальными формами диабета, отнесенными к «другим специфическим формам диабета», генез патологии почек будет связан с диабетическими нарушениями, поскольку эти пациенты характеризуются крайними формами инсулинорезистентности и могут иметь тяжелую гипергликемию в течение длительного периода. Однако классическая ДН у пациентов с наследственными липодистрофиями встречается редко, но у них наблюдаются другие заболевания почек [35].
Согласно данным литературы, при обследовании пациентов с ВГЛ или ПГЛ часто диагностируются хронические заболевания почек, проявляющиеся протеинурией, среди которых наиболее распространены фокальный сегментарный гломерулосклероз (ФСГС) и мембрано-пролиферативный гломерулонефритит (МПГН). С другой стороны, связь между СПЛ и заболеваниями почек была задокументирована в очень немногих случаях [36].
Эти данные согласуются с результатами исследования Javor E. и соавт. [37], в котором были обследованы 25 пациентов с ГЛ (7 с ПГЛ и 18 с ВГЛ). У 88% была диагностирована микроальбуминурия (>30 мг/сут), у 60% — макроальбуминурия (>300 мг/сут) и у 20% — протеинурия нефротического диапазона (>3500 мг/сут). У 23 (92%) пациентов наблюдался повышенный клиренс креатинина (>125 мл/мин/1,73 м²). 7 из 25 обследованных пациентов была проведена биопсия почек, по результатам которой у 4 пациентов был диагностирован ФСГС, у 2 — МПГН и у 1 пациента — признаки ДН.
Патогенез поражения почек при наследственных липодистрофиях остается неясным. Возможная патогенная связь между мутациями LMNA и возникновением ФСГС связана с трансформирующим фактором роста-бета1 (TGF-β1). У трансгенных мышей, сверхэкспрессирующих активный человеческий TGF-β1, развивается фиброз печени, а также гломерулосклероз и синдром, подобный липодистрофии. Сверхэкспрессия TGF-β1 в подоцитах может привести к подоцитопении и гломерулосклерозу. Было высказано предположение, что ламины А-типа необходимы для ингибирования пролиферации фибробластов путем контроля активации генов, стимулируемых TGF-β1 [38–41].
Специфические изменения внешнего вида и фигуры, обусловленные липодистрофией, почти всегда являются причиной социальной стигматизации пациентов и одной из ведущих причин для обращения за психологической помощью.
В исследовании, проведенном Ajluni N. и соавт. [17] с участием 22 пациентов с СПЛ и одним пациентом с ППЛ, расстройство настроения, требующее приема лекарств, было выявлено у 12 пациентов (52,2%). Депрессия была наиболее распространенным психиатрическим заболеванием, присутствующим у 10 пациентов (43,5%). У шести пациентов была выявлена тревожность (26,1%), у одного пациента было биполярное расстройство, а еще у одного было неустановленное психиатрическое заболевание, для лечения которого он ранее принимал антипсихотическое средство. Хроническая боль присутствовала у 78,3% пациентов и, как сообщается, была вызвана сочетанием артрита, болей в спине, фибромиалгии и миопатии.
Подобные нарушения могут приводит к крайне серьезным последствиям, в том числе к попыткам суицида, в связи с чем своевременная психологическая и психиатрическая помощь имеет важное значение для этих пациентов.
Основными причинами смерти пациентов с наследственными липодистрофиями являются заболевания сердца (кардиомиопатия, сердечная недостаточность, инфаркт миокарда, аритмия), печени (печеночная недостаточность, желудочно-кишечные кровотечения, гепатоцеллюлярная карцинома), почечная недостаточность, острый панкреатит и сепсис [5][9].
В исследовании Lima J. и соавт. из 20 пациентов (12 женщин и 8 мужчин), умерших от ВГЛ в период с 1997 по 2017 гг., средний возраст на момент смерти составил 27,1±12,4 года. Возраст смерти женщин был более молодой, чем у мужчин (25,2±12,5 против 29,9±12,6 года, р=0,41). Ожидаемая продолжительность жизни исследуемой популяции составила 62,9±4,8 года. Авторы представили классификацию причин смерти, разделив их на 3 основные группы: инфекции, заболевания печени и прочие причины (включая острый панкреатит, почечную недостаточность и внезапную смерть/инфаркт миокарда). У 7 человек (35%) была зарегистрирована смерть из-за инфекционного процесса. Наиболее распространенной причиной в этой категории стала пневмония, которая развилась у четырех пациентов. У двоих наблюдалась дыхательная недостаточность. Один пациент из инфекционной группы скончался от целлюлита правого бедра, возникшего на фоне неполного разрыва мышцы и септического артрита. Еще семь пациентов (35%) умерли от заболеваний печени или их осложнений. Трое из них погибли от кровотечения из верхних отделов желудочно-кишечного тракта, вызванного циррозом печени. Четыре пациента имели тяжелые заболевания печени и скончались от печеночной недостаточности. Два пациента умерли внезапно; по данным вскрытия одного из них, был диагностирован острый инфаркт миокарда. Один пациент скончался от острого панкреатита, причиной смерти еще двоих стала почечная недостаточность [42].
Gupta N. и соавт. исследовали причины смерти пациентов с ВГЛ, диагностированным в детском возрасте. Из 15 участников исследования 6 (40%) скончались от респираторной инфекции, а 1 — от перитонита [42]. Rheuban KS. и соавт. описали клинический случай смерти пациента от перитонита и септицемии в возрасте 23 лет [43]. Две из четырех зарегистрированных смертей, о которых сообщали Bjornstad PG. и соавт., также были связаны с инфекциями легких; средний возраст на момент смерти составил 32 года [44]. Hsu RH. и соавт. представили данные о 16 педиатрических пациентах с ВГЛ типа 2 в Тайване. Трое из этих пациентов скончались в среднем в возрасте 10,9 года: один от аритмии, другой — от двусторонней пневмонии, а третий — по неизвестной причине [45].
Эти данные указывают на то, что инфекция, чаще легочная, является одной из основных причин смерти. Что касается возможных механизмов, объясняющих высокую предрасположенность к инфекционным процессам в данной группе, то на макрофагах присутствуют рецепторы лептина, который активирует фагоцитарную функцию этих клеток. Низкие уровни лептина в сыворотке у пациентов с ВГЛ могут способствовать развитию и прогрессированию инфекционных процессов. Также возможными механизмами могут быть нарушения в мембранных липидах или липидных рафтах, что может облегчать проникновение внутриклеточных паразитов или приводить к дефектной презентации антигена. Кроме того, уровень холестерина ЛПВП ниже 0,35 ммоль/л ассоциируется с повышенной предрасположенностью к инфекциям [46].
Диагностика наследственных липодистрофий у пациентов осложняется многообразием системных проявлений заболевания и недостаточной осведомленностью врачей общего профиля. Обучение клиницистов систематическому включению анализа жировой ткани в стандартную клиническую практику должно способствовать улучшению диагностики и терапии липодистрофии, а также сопутствующих заболеваний, связанных с этими синдромами [13].
Очевидны негативные последствия задержки в установлении диагноза для здоровья пациента, прогноза заболевания и затрат на лечение. Среди врачей общего профиля, а особенно гастроэнтерологов, гинекологов, кардиологов и терапевтов, важно повысить осведомленность о первичной диагностике липодистрофии, чтобы обеспечить раннее установление диагноза и своевременное лечение.
Источник финансирования. Финансирование данной работы не проводилось.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Участие авторов. Фролкова Н.В. — анализ медицинской документации пациента и написание текста; Кокшарова Е.О. — анализ литературных данных, окончательное утверждение для публикации рукописи; Мишина Е.Е. — анализ литературных данных, редактирование текста и утверждение для публикации рукописи; Шестакова М.В. — разработка концепции и дизайна, анализ литературных данных, окончательное редактирование текста и утверждение для публикации рукописи.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
1. Hussain I, Patni N, Garg A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology. 2019;51(2):202-212. doi: https://doi.org/10.1016/j.pathol.2018.11.004
2. Akinci B, Oral EA, Neidert A, et al. Comorbidities and Survival in Patients With Lipodystrophy: An International Chart Review Study. J Clin Endocrinol Metab. 2019;104(11):5120-5135. doi: https://doi.org/10.1210/jc.2018-02730
3. Cook K, Adamski K, Gomes A, et al. Effects of Metreleptin on Patient Outcomes and Quality of Life in Generalized and Partial Lipodystrophy. J Endocr Soc. 2021;5(4):bvab019. doi: https://doi.org/10.1210/jendso/bvab019
4. Patni N, Garg A. Lipodystrophy for the Diabetologist-What to Look For. Curr Diab Rep. 2022;22(9):461-470. doi: https://doi.org/10.1007/s11892-022-01485-w
5. Brown RJ, Araujo-Vilar D, Cheung PT, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016;101(12):4500-4511. doi: https://doi.org/10.1210/jc.2016-2466
6. Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of lipodystrophy - an AACE consensus statement. Endocr Pract. 2013;19(1):107-116. doi: https://doi.org/10.4158/endp.19.1.v767575m65p5mr06
7. Rêgo AG, Mesquita ET, Faria CA, et al. Anormalidades cardiovasculares e metabólicas em pacientes com a síndrome de Berardinelli-Seip [Cardiometabolic abnormalities in patients with Berardinelli-Seip syndrome]. Arq Bras Cardiol. 2010;94(1):109-118. doi: https://doi.org/10.1590/s0066-782x2010000100017
8. Patni N, Garg A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction. Nat Rev Endocrinol. 2015;11(9):522-534. doi: https://doi.org/10.1038/nrendo.2015.123
9. Khalife WI, Mourtada MC, Khalil J. Dilated cardiomyopathy and myocardial infarction secondary to congenital generalized lipodystrophy. Tex Heart Inst J. 2008;35(2):196-199
10. Соркина Е.Л., Тюльпаков А.Н. Наследственные и приобретенные липодистрофии: молекулярно-генетические и аутоиммунные механизмы // Ожирение и метаболизм. — 2018. — Т. 15. — №1. — С. 39-42. https://doi.org/10.14341/omet2018139-42
11. Lazarte J, Wang J, McIntyre AD, Hegele RA. Prevalence of severe hypertriglyceridemia and pancreatitis in familial partial lipodystrophy type 2. J Clin Lipidol. 2021;15(5):653-657. doi: https://doi.org/10.1016/j.jacl.2021.07.004
12. Chan D, McIntyre AD, Hegele RA, Don-Wauchope AC. Familial partial lipodystrophy presenting as metabolic syndrome. J Clin Lipidol. 2016;10(6):1488-1491. doi: https://doi.org/10.1016/j.jacl.2016.08.012
13. Mosbah H, Vatier C, Andriss B, et al. Patients’ perspective on the medical pathway from first symptoms to diagnosis in genetic lipodystrophy. Eur J Endocrinol. 2024;190(1):23-33. doi: https://doi.org/10.1093/ejendo/lvad169
14. Летова Е.К., Старкова Н.Т. Дифференциальная диагностика синдрома генерализованной липодистрофии // Проблемы Эндокринологии. — 1993. — Т. 39. — №5. — С. 33-36. doi: https://doi.org/10.14341/probl11960
15. Huang-Doran I, Sleigh A, Rochford JJ, et al. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol. 2010;207(3):245-255. doi: https://doi.org/10.1677/JOE-10-0272
16. Bindlish S, Presswala LS, Schwartz F. Lipodystrophy: Syndrome of severe insulin resistance. Postgrad Med. 2015;127(5):511-516. doi: https://doi.org/10.1080/00325481.2015.1015927
17. Ajluni N, Meral R, Neidert AH, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf). 2017;86(5):698-707. doi: https://doi.org/10.1111/cen.13311
18. Fourman LT, Lima JG, Simha V, et al. A rapid action plan to improve diagnosis and management of lipodystrophy syndromes. Front Endocrinol (Lausanne). 2024;15:1383318. doi: https://doi.org/10.3389/fendo.2024.1383318
19. Safar Zadeh E, Lungu AO, Cochran EK, et al. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J Hepatol. 2013;59(1):131-137. doi: https://doi.org/10.1016/j.jhep.2013.02.007
20. Lüdtke A, Genschel J, Brabant G, et al. Hepatic steatosis in Dunnigan-type familial partial lipodystrophy. Am J Gastroenterol. 2005;100(10):2218-2224. doi: https://doi.org/10.1111/j.1572-0241.2005.00234.x
21. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20(5):649-688. doi: https://doi.org/10.1210/edrv.20.5.0380
22. Bjørnstad PG, Foerster A, Ihlen H. Cardiac findings in generalized lipodystrophy. Acta Paediatr Suppl. 1996;413:39-43. doi: https://doi.org/10.1111/j.1651-2227.1996.tb14264.x
23. Debray FG, Baguette C, Colinet S, et al. Early infantile cardiomyopathy and liver disease: a multisystemic disorder caused by congenital lipodystrophy. Mol Genet Metab. 2013;109(2):227-229. doi: https://doi.org/10.1016/j.ymgme.2013.04.011
24. Van Maldergem L, Magré J, Khallouf TE, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy [published correction appears in J Med Genet. 2003 Feb;40(2):150.]. J Med Genet. 2002;39(10):722-733. doi: https://doi.org/10.1136/jmg.39.10.722
25. Lupsa BC, Sachdev V, Lungu AO, et al. Cardiomyopathy in congenital and acquired generalized lipodystrophy: a clinical assessment. Medicine (Baltimore). 2010;89(4):245-250. doi: https://doi.org/10.1097/MD.0b013e3181e9442f
26. Akinci B, Onay H, Demir T, et al. Natural History of Congenital Generalized Lipodystrophy: A Nationwide Study From Turkey. J Clin Endocrinol Metab. 2016;101(7):2759-2767. doi: https://doi.org/10.1210/jc.2016-1005
27. Andre P, Schneebeli S, Vigouroux C, et al. Metabolic and cardiac phenotype characterization in 37 atypical Dunnigan patients with nonfarnesylated mutated prelamin A. Am Heart J. 2015;169(4):587-593. doi: https://doi.org/10.1016/j.ahj.2014.12.021
28. Hegele RA. Premature atherosclerosis associated with monogenic insulin resistance. Circulation. 2001;103(18):2225-2229. doi: https://doi.org/10.1161/01.cir.103.18.2225
29. Decaudain A, Vantyghem MC, Guerci B, et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab. 2007;92(12):4835-4844. doi: https://doi.org/10.1210/jc.2007-0654
30. Lungu AO, Zadeh ES, Goodling A, et al. Insulin Resistance Is a Sufficient Basis for Hyperandrogenism in Lipodystrophic Women With Polycystic Ovarian Syndrome. Obstetrical & Gynecological Survey. 2012;67(9):549-550. doi: https://doi.org/10.1097/01.ogx.0000421454.21771.e8
31. Upreti V, Dhull P, Patnaik SK, Kumar KV. An unusual cause of delayed puberty: Berardinelli- Seip syndrome. J Pediatr Endocrinol Metab. 2012;25(11-12):1157-1160. doi: https://doi.org/10.1515/jpem-2012-0240
32. Fernandez-Pombo A, Diaz-Lopez EJ, Castro AI, et al. Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review. Cells. 2023;12(5):725. doi: https://doi.org/10.3390/cells12050725
33. Akinci B, Onay H, Demir T, et al. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy. Metabolism. 2017;72:109-119. doi: https://doi.org/10.1016/j.metabol.2017.04.010
34. Vantyghem MC, Vincent-Desplanques D, Defrance-Faivre F, et al. Fertility and obstetrical complications in women with LMNA-related familial partial lipodystrophy. J Clin Endocrinol Metab. 2008;93(6):2223-2229. doi: https://doi.org/10.1210/jc.2007-2521
35. Musso C, Javor E, Cochran E, et al. Spectrum of renal diseases associated with extreme forms of insulin resistance. Clin J Am Soc Nephrol. 2006;1(4):616-622. doi: https://doi.org/10.2215/CJN.01271005
36. Fountas A, Giotaki Z, Dounousi E, et al. Familial partial lipodystrophy and proteinuric renal disease due to a missense c.1045C > T LMNA mutation. Endocrinol Diabetes Metab Case Rep. 2017;2017:17-0049. doi: https://doi.org/10.1530/EDM-17-0049
37. Javor ED, Moran SA, Young JR, et al. Proteinuric nephropathy in acquired and congenital generalized lipodystrophy: baseline characteristics and course during recombinant leptin therapy. J Clin Endocrinol Metab. 2004;89(7):3199-3207. doi: https://doi.org/10.1210/jc.2003-032140
38. Thong KM, Xu Y, Cook J, et al. Cosegregation of focal segmental glomerulosclerosis in a family with familial partial lipodystrophy due to a mutation in LMNA. Nephron Clin Pract. 2013;124(1-2):31-37. doi: https://doi.org/10.1159/000354716
39. Clouthier DE, Comerford SA, Hammer RE. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Investig. 1997;100(11):2697–713. doi: https://doi.org/10.1172/jci119815
40. Van Berlo JH, Voncken JW, Kubben N, et al. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet. 2005;14(19):2839-2849. doi: https://doi.org/10.1093/hmg/ddi316
41. Lee HS, Song CY. Effects of TGF-beta on podocyte growth and disease progression in proliferative podocytopathies. Kidney Blood Press Res. 2010;33(1):24-29. doi: https://doi.org/10.1159/000285844
42. Gupta N, Asi N, Farah W, et al. Clinical Features and Management of Non-HIV-Related Lipodystrophy in Children: A Systematic Review. J Clin Endocrinol Metab. 2017; 102(2):363–74. doi: https://doi.org/10.1210/jc.2016-2271
43. Rheuban KS, Blizzard RM, Parker MA, et al. Hypertrophic cardiomyopathy in total lipodystrophy. J Pediatr. 1986;109(2):301–2.
44. Bjornstad PG, Foerster A, Ihlen H. Cardiac findings in generalized lipodystrophy. Acta Paediatr Suppl. 1996;413:39–43.
45. Hsu RH, Lin WD, Chao MC, et al. Congenital generalized lipodystrophy in Taiwan. J Formos Med Assoc. 2018;118(1 Pt 1):142-147. doi: https://doi.org/10.1016/j.jfma.2018.02.003
46. Lima JG, Nobrega LHC, Lima NN, et al. Causes of death in patients with Berardinelli-Seip congenital generalized lipodystrophy. PLoS One. 2018;13(6):e0199052. doi: https://doi.org/10.1371/journal.pone.0199052
Фролкова Надежда Викторовна, аспирант.
117036, Москва, ул. Дм. Ульянова, д. 11
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи
Кокшарова Екатерина Олеговна - н.с.
Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи
Мишина Екатерина Евгеньевна - к.м.н., н.с.
Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи
Шестакова Марина Владимировна - д.м.н., профессор, академик РАН.
Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи
|
|
1. Рисунок 1. Фенотип пациенток с различными формами наследственных липодистрофий (из архива ГНЦ РФ ФГБУ «НМИЦ эндокринологии им. академика И.И. Дедова» Минздрава России). На 1 и 2 фотографиях — пациентки с генерализованными липодистрофиями, фотография 3 — атипичный прогероидный синдром (мутация в гене LMNA), фотографии 4 и 5 — пациентки с семейными парциальными липодистрофиями, фотография 6 — acanthosis nigricans подмышечной впадины. Красной стрелкой обозначены зоны липогипертрофии; черной — зоны липодистрофии. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(1MB)
|
Метаданные ▾ | |
Фролкова Н.В., Кокшарова Е.О., Мишина Е.Е., Шестакова М.В. Наследственные липодистрофии: как не пропустить диагноз? Сахарный диабет. 2025;28(4):413-423. https://doi.org/10.14341/DM13314
Frolkova N.V., Koksharova E.O., Mishina E.E., Shestakova M.V. Hereditary lipodystrophies: how not to miss the diagnosis? Diabetes mellitus. 2025;28(4):413-423. (In Russ.) https://doi.org/10.14341/DM13314

 |
Адрес: 117036, Российская Федерация, Москва, улица Дмитрия Ульянова, дом 11.
Обработка персональных данных







































