




Содержание
Перейти к:
https://doi.org/10.14341/DM13416
Перейти к:
Интерес к изучению продуктов промежуточного метаболизма и клеточной биоэнергетики связан с глобальным ростом распространенности метаболических заболеваний. Структурные особенности костной ткани с повышенным риском низкоэнергетических переломов при сахарном диабете связаны с комплексными нарушениями костного метаболизма, включая накопление конечных продуктов гликирования, подавление остеобластогенеза, усиление экспрессии склеростина, развитие окислительного стресса и усиление катаболических процессов в клетках под влиянием гипергликемии. Патогенетические изменения при остеопорозе в свою очередь связаны со сложными изменениями клеточного энергообмена, митохондриальной функции, а также нарушениями синтеза и распада пуринов, которые способствуют прогрессирующему дисбалансу костного ремоделирования и снижению биомеханических свойств костной ткани. В настоящем обзоре литературы собрана информация об основных источниках и путях синтеза аденозинтрифосфата в костных клетках, механизмах регуляции энергообмена в норме, а также в условиях гипергликемии при сахарном диабете и в условиях возрастных изменений организма с развитием остеопороза. Представленные данные открывают новые перспективы для разработки таргетной терапии описанных метаболических нарушений как потенциального подхода к профилактике и лечению патологии костной ткани.
Жданова А.С., Белая Ж.Е., Катаева Д.А., Омельченко К.А. Изменения энергетического обмена костной ткани при сахарном диабете и старении как причина повышенной хрупкости скелета. Сахарный диабет. 2026;29(2):191-202. https://doi.org/10.14341/DM13416
Zhdanova A.S., Belaya Zh.E., Kataeva D.A., Omelchenko K.A. Changes in bone tissue energy metabolism in diabetes mellitus and aging as a cause of increased skeletal fragility. Diabetes mellitus. 2026;29(2):191-202. (In Russ.) https://doi.org/10.14341/DM13416
Энергетический обмен является неотъемлемой частью любой клетки организма для обеспечения процессов ее эффективного функционирования, поддержания гомеостаза и реагирования на изменения внешней среды. Клетки, ответственные за костный метаболизм — остеобласты и остеокласты, — требуют значительных количеств энергетических субстратов (глюкозы, жирных кислот, глутамина) для обеспечения процессов дифференцировки и выполнения специфических функций. Современные данные свидетельствуют о тесной взаимосвязи ключевых сигнальных путей, регулирующих развитие костных клеток, с путями энергетического обмена, способными динамически адаптироваться к изменяющимся энергетическим потребностям на разных этапах клеточного цикла. Основными метаболическими путями синтеза аденозинтрифосфата (АТФ) являются процесс гликолиза, цикл трикарбоновых кислот (цикл Кребса), окислительное фосфорилирование, а также окисление жирных кислот и метаболизм глутамина [1].
Многообещающим направлением для понимания путей патогенеза метаболических заболеваний скелета становится изучение особенностей энергообмена в клетках костной системы, в частности в условиях нарушения обмена глюкозы при сахарном диабете (СД), а также в условиях естественного старения с развитием первичного остеопороза, что может усугубляться нарушениями углеводного обмена.
До XXI века костную ткань относили к органам, функционирующим в условиях гипоксии и получающим энергию преимущественно при помощи путей гликолиза. Однако с развитием новых технологий оценки васкуляризации был сформулирован вывод о том, что остеобласты и остеоциты имеют достаточное снабжение кислородом, так как получают кровоснабжение из сосудов кости, а не из костного мозга, в связи с чем могут получать энергию в том числе в аэробных условиях, используя процесс цикла Кребса и окислительного фосфорилирования в митохондриях [2].
При этом клетки остеогенной линии пластичны и могут использовать различные виды топлива и энергетические пути в зависимости от их статуса дифференцировки, окружающей среды и потребностей тканей [2].
Остеобластогенез начинается с мезенхимальных стволовых клеток костного мозга, которые также являются предшественниками для синтеза адипоцитов. Помимо образования клеток костного матрикса, мезенхимальные стволовые клетки в процессе метаболизма костной ткани способны уменьшать созревание и активацию предшественников остеокластов, регулируя активность тартрат-резистентной кислой щелочной фосфатазы, матриксной металлопептидазы и катепсина К [3].
По мере пролиферации и дифференцировки клетки остеогенной линии (остеобласты, остеоциты) переходят от гликолитического пути на этапе пролиферации к увеличению активности окислительного фосфорилирования в качестве источника энергии для функционирования зрелых клеток (рис. 1) [2].
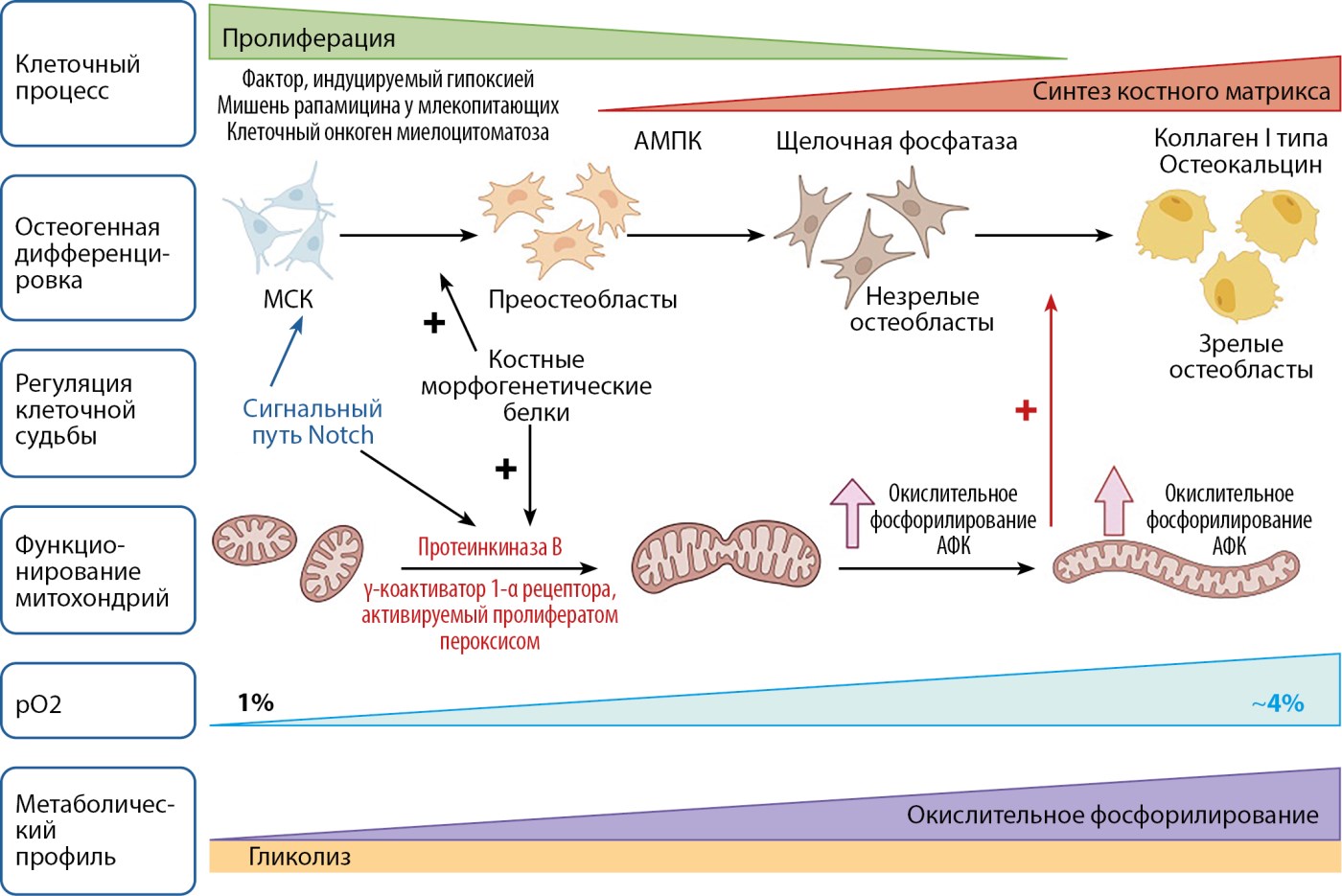
Рисунок 1. Биоэнергетическая регуляция и сигнальные пути в процессе дифференцировки остеобластов (создано при помощи BioRender).
Примечание. В процессе дифференцировки остеобласты в качестве источника энергии переходят от гликолиза к окислительному фосфорилированию. Преобладание гликолиза у преостеобластов обусловлено наличием в клетках незрелых митохондрий, а также повышенной экспрессией фактора, индуцируемого гипоксией под влиянием сигнального пути Notch, который ингибирует окислительное фосфорилирование и активирует гликолиз.
Созревание остеобластов сопровождается метаболическим сдвигом в сторону большего потребления кислорода, увеличением количества крипт митохондрий с активацией окислительного фосфорилирования за счет действия костных морфогенетических белков.
Notch — семейство трансмембранных белков; АФК — активные формы кислорода; AМПК — АМФ-активируемая протеинкиназа; МСК — мультипотентные стволовые клетки.
Важным регулятором в выборе пути биосинтеза энергии в процессе созревания мультипотентных стволовых клеток выступает сигнальный путь Notch. Действие Notch реализуется через HIF (Hypoxia-Inducible Factor — фактор, индуцируемый гипоксией), увеличивая его экспрессию, что приводит к усилению гликолиза в клетках. Однако последние работы публикуют данные о подавлении митохондриального и гликолитического метаболизма аномально повышенной передачей сигналов Notch [2].
Использование преимущественно гликолиза низкодифференцированными преостеобластами объясняется наличием в клетках фрагментированных органелл с незрелыми криптами митохондрий, не способными к адекватному окислительному фосфорилированию, а также обеспечение гликолизом поддержки низкого уровня активных форм кислорода, сохраняя возможность предотвращать повреждение генетического материала клеток. Эти данные согласуются с публикациями об эмбриональных стволовых клетках и соматических стволовых клетках из других тканей [2].
Потребность клеток в энергии на различных этапах их созревания реализуется через увеличение экспрессии GLUT-1, регулирование гликолиза и окислительного фосфорилирования HIF, каскадом сигнального пути Wnt/β-катенин, костными морфогенетическими белками (BMP), а также системой передачи сигналов внутри клетки АМФ-активируемой протеинкиназой (АМПК) и мишенью рапамицина у млекопитающих (mTOR).
Вклад HIF в регуляцию биоэнергетики преимущественно заключается в подавлении окислительного фосфорилирования и одновременной активации гликолиза. Фактор, индуцируемый гипоксией, усиливает захват глюкозы, снижает окисление пирувата в митохондриях за счет повышения экспрессии пируватдегидрогеназкиназы, а также дополнительно стимулирует транскрипцию ключевых гликолитических ферментов, таких как лактатдегидрогеназа, что приводит к восстановлению пирувата в цикле трикарбоновых кислот до лактата. Параллельно HIF подавляет β-окисление жирных кислот, еще больше ограничивая митохондриальный энергетический метаболизм [4].
Сигнальный каскад Wnt/β-катенин представляет собой важный регуляторный механизм, управляющий остеогенной дифференцировкой. Данный путь инициирует активацию мультипотентных мезенхимальных стволовых клеток, стимулируя их пролиферацию и запуская ранние этапы остеогенеза [5]. Согласно имеющимся данным, передача сигналов Wnt способна потенцировать как гликолитические процессы, так и окислительное фосфорилирование, причем выраженность эффекта определяется продолжительностью воздействия. Однако существенным методологическим ограничением большинства исследований в данной области является применение иммортализованных клеточных линий вместо первичных культур, поскольку процесс иммортализации — бесконечного деления — существенно влияет на метаболическое программирование клеток [6].
Костные морфогенетические белки регулируют созревание и дифференцировку костных клеток активируя экспрессию ключевых генов (Runx2-фактора транскрипции дифференцировки остеобластов), а также воздействуя через неканонические (Akt/mTOR/MAPK) механизмы. Однако данные об их влиянии на биоэнергетику противоречивы: Smith CO и Eliseev RA в 2021 г. описали активацию окислительного фосфорилирования под действием BMP с незначительным влиянием на гликолиз [7]. В то же время накоплены данные о стимулировании BMP в отношении mTOR и HIF1α, что позволяет предполагать воздействие BMP на гликолитический метаболизм в остеогенной линии [8][9].
Клетки остеогенной линии в качестве инструментов адаптации обладают сложными системами восприятия метаболитов и энергии, такими как АМФ-активируемая протеинкиназа (АМПК) и мишень рапамицина у млекопитающих (mTOR). АМФ-активируемая протеинкиназа выполняет функцию детектора уровня глюкозы и общего энергетического баланса клетки, тогда как mTOR служит главным рецептором доступности аминокислот. Активация AMПK стимулирует процессы окислительного фосфорилирования при помощи двух путей: 1) усиливает митохондриальный биогенез, увеличивая количество митохондрий; 2) потенцирует экспрессию костного морфогенетического белка 2 (BMP2), который индуцирует окислительное фосфорилирование. Роль mTOR в остеогенезе заключается в регуляции пролиферации преостеобластов путем стимулирования гликолиза через активацию транскрипционных факторов HIF1α и c-Myc, тогда для терминальной дифференцировки и окончательного созревания остеобластов необходима его инактивация. В то же время сигнальный путь mTOR контролирует активность АМФ-активируемой протеинкиназы [10][11].
Дифференцировка и созревание клеток проявляется метаболическим сдвигом в сторону большего потребления кислорода, увеличения количества крипт митохондрий с активацией окислительного фосфорилирования и образованием активных форм кислорода, синтеза лактата.
Остеокласты в свою очередь происходят из клеток линии моноцитов/макрофагов, которые, сливаясь в многоклеточные гигантские клетки, способны к резорбции костной ткани. Процесс дифференцировки остеокластов требует метаболического перепрограммирования для поддержания биосинтетических субстратов и энергоснабжения (рис. 2) [12].
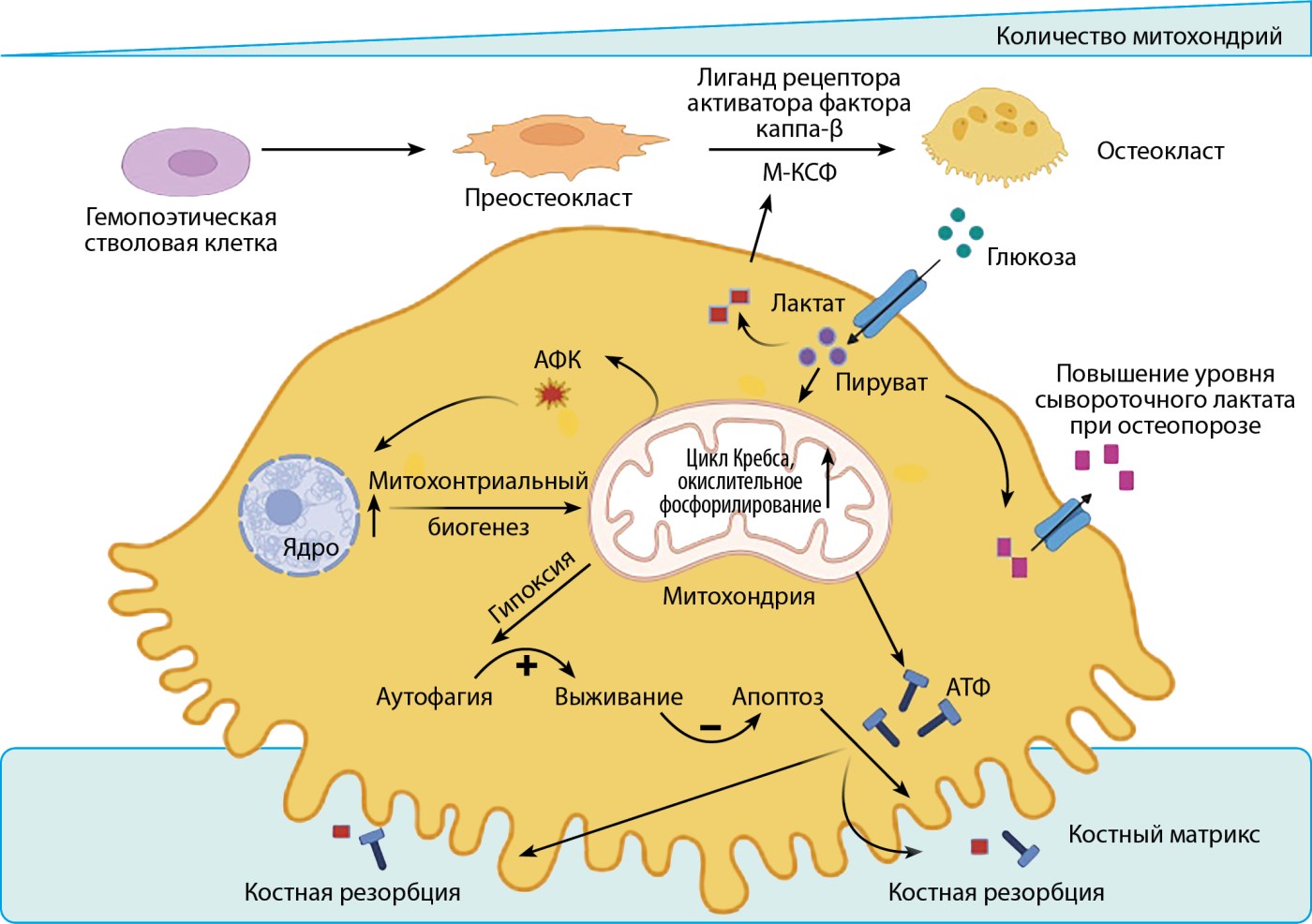
Рисунок 2. Особенности энергетического обмена в остеокласте в процессе остеокластогенеза (создано при помощи BioRender).
Примечание. В основе остеопороза лежит повышение активности остеокластов. Высокая потребность в энергии в процессе дифференцировки остеокластов обеспечивается путем увеличения количества митохондрий в клетках, усиления экспрессии ферментов, участвующих в окислительном фосфорилировании и цикле Кребса. Повышение деградации костного матрикса при остеопорозе поддерживается путем анаэробного гликолиза в условиях гипоксии, приводящего к накоплению лактата. Дополнительным механизмом поддержания энергообмена является аутофагия в остеокластах. АТФ — аденозинтрифосфат; М-КСФ — колониестимулирующий фактор макрофагов; цикл Кребса — цикл трикарбоновых кислот; АФК — активные формы кислорода.
Остеокластогенез осуществляется благодаря значительному увеличению поглощения глюкозы предшественниками остеокластов за счет транспортеров GLUT-1 и GLUT-3, возрастания количества митохондрий в клетках, повышения скорости потребления кислорода и усилением экспрессии ферментов, участвующих в гликолизе, цикле трикарбоновых кислот и окислительном фосфорилировании. Кроме того, хотя дифференцировка остеокластов в основном зависит от глюкозы как источника энергии, пороговый уровень еще не установлен (слишком высокий или слишком низкий уровень глюкозы, вероятно, негативно влияет на активность резорбции костной ткани) [12].
Функция остеокластов реализуется при создании кислой среды ионами водорода H+ с помощью карбоангидразы II, что способствует формированию локального ацидоза для разрушения неорганических минералов кости [12].
В работе Taubmann J и соавт. была выдвинута гипотеза об усилении гликолиза и выработке лактата в анаэробных условиях в случае резорбции костной ткани. При фармакологическом блокировании гликолиза с использованием 2-дезокси-D-глюкозы эффективно происходило ингибирование резорбции кости как в условиях in vitro, так и in vivo, однако не оказывало влияния на дифференцировку остеокластов и их жизнеспособность. Таким образом, необходимая энергия для дифференцировки остеокластов в основном вырабатывается в процессе окислительного фосфорилирования, а деградация костного матрикса остеокластами поддерживается гликолитическим процессом [13].
Дополнительным источником синтеза энергии в остеокластах выступает образование АТФ при окислении жирных кислот. Однако в условиях гипоксии остеокласты претерпевают изменения в виде метаболической адаптации для сохранения своего функционирования, что проявляется накоплением липидных включений и нарушением утилизации жирных кислот. Компенсаторно активируется HIF-зависимый захват глутамина, который служит источником α-кетоглутарата для цикла трикарбоновых кислот [14].
Особенности вторичного остеопороза и изменения энергетического обмена костной ткани в условиях нарушения метаболизма глюкозы при сахарном диабете
Пациенты с СД имеют клинические особенности течения остеопороза и повышенный риск переломов при сравнении с пациентами без нарушений углеводного обмена, что связано со снижением скорости обновления костной ткани и нарушением ее микроархитектоники (рис. 3) [15].
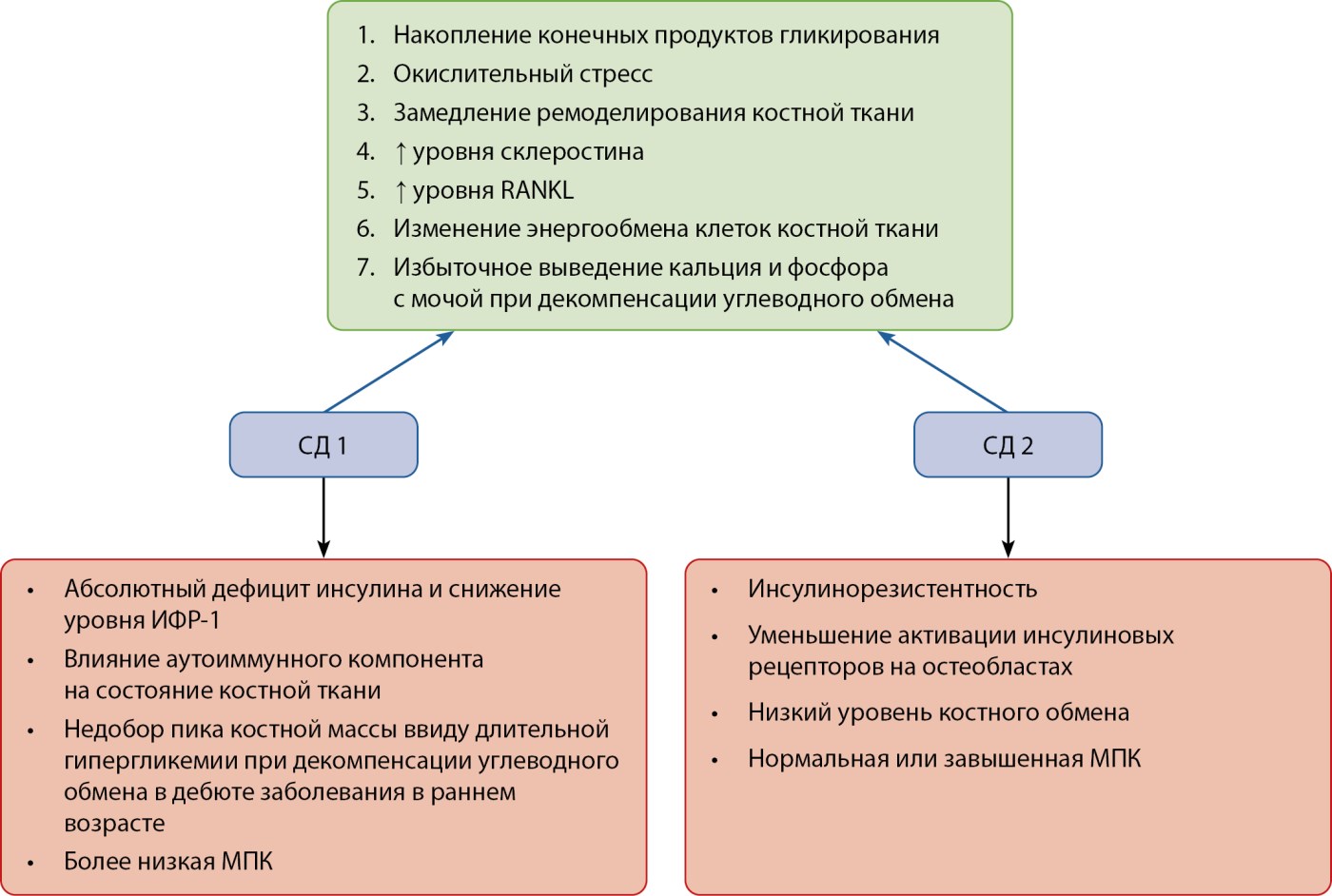
Рисунок 3. Общие и отличные патогенетические механизмы нарушения костного ремоделирования при сахарном диабете 1 и 2 типа.
Примечание. Гипергликемия при сахарном диабете 1 и 2 типа приводит к накоплению конечных продуктов гликирования, окислительному стрессу, повышению уровня RANKL и повышению уровня склеростина, а также избыточному выведению кальция и фосфора почками при длительной декомпенсации углеводного обмена, что замедляет и ухудшает качество ремоделирования костной ткани. Абсолютный дефицит инсулина при сахарном диабете 1 типа ассоциирован с низким уровнем ИФР-1 и его сниженным анаболическим влиянием на костную ткань. Кроме того, для пациентов с сахарным диабетом 1 типа характерен недобор пика костной массы в молодом возрасте ввиду раннего дебюта заболевания и влияния декомпенсации углеводного обмена, что клинически характеризуется более низкой МПК. В условиях сахарного диабета 2 типа вследствие инсулинорезистентности уменьшается активация инсулиновых рецепторов на остеобластах, что снижает привлечение остеокластов и их функциональную активность. Низкий уровень костного обмена обуславливает наличие нормальной или завышенной МПК у данных пациентов.
В ряде отечественных исследований показано, что при СД изменения костного обмена обусловлены несколькими факторами: дефицитом или наличием дефектного инсулина, накоплением конечных продуктов гликирования (КПГ), хроническим воспалением, оксидативным стрессом, влиянием осложнений СД: микроангиопатий, вторичного гиперпаратиреоза на фоне хронической болезни почек [16].
Отдельную роль играет плохой гликемический контроль у пациентов с СД, что подавляет экспрессию Runx2 (связанный с Runt транскрипционный фактор-2), участвующего в остеобластогенезе [1].
Основным субстратом в скелете для формирования конечных продуктов гликирования (КПГ) является коллаген 1‑го типа, представляющий порядка 90% органического компонента костной ткани. Активация рецепторов к КПГ приводит к образованию активных форм кислорода и провоспалительных цитокинов, что поддерживает хроническое воспаление при СД [15].
Неферментное гликирование коллагена приводит к формированию поперечных сшивок в коллагене, которые уменьшают пластичность костной ткани и снижают устойчивость к механическому воздействию, что в сочетании с повышенным риском падений приводит к появлению переломов [15].
Farlay D. были проанализированы данные биопсии подвздошной кости у пациентов с СД 1 типа (СД1) с низкотравматическими переломами и без, после чего результаты сравнили с данными здоровых людей без СД и без переломов (n=5 человек в каждой группе). Авторы оценивали содержание основного конечного продукта гликирования — пентозидина в кортикальной и трабекулярной костной ткани. У пациентов с СД1 содержание пентозидина в трабекулярной костной ткани было значительно выше и положительно коррелировало с уровнем гликированного гемоглобина [17].
T. Neumann и соавт. оценили взаимосвязь уровня пентазидина в сыворотке крови у пациентов с СД1 в пременопаузе (мужчины и женщины n=128) с частотой переломов и степенью снижения минеральной плотности кости в сравнении с контрольной группой. По данным работы, уровень пентозидина у пациентов с СД1 являлся независимым фактором возникновения патологических переломов, вне зависимости от минеральной плотности кости [18].
Гипергликемия и ее конечные продукты подавляют дифференцировку остеобластов и образование новой костной ткани напрямую и опосредованно путем увеличения экспрессии склеростина в остеоцитах [19][20].
Склеростин представляет собой гликопротеин, секретируемый остеоцитами, который выступает в роли антагониста сигнального пути Wnt. В норме активация пути Wnt стимулирует дифференцировку остеобластогенеза из мезенхимальных клеток-предшественников и снижение их апоптоза. Склеростин, связываясь конкурентно с LRP-5 (трансмембранный рецептор липопротеинов низкой плотности), напротив, приводит к снижению скорости образования кости [5][15][21].
Antonia García-Martín и соавт. продемонстрировали результаты исследования по оценке уровня склеростина у пациентов с СД2 (СД2) (n=74, контроль n=50). Уровень циркулирующего склеростина был повышен при СД2 независимо от пола и возраста, при этом у пациентов с остеопорозом и СД показатели были выше, чем у пациентов с СД без остеопороза. Была выявлена корреляция его уровня со значением гликированного гемоглобина (HbA1c), маркерами костного обмена, минеральной плотностью кости, а также с продолжительностью заболевания СД2 [22].
В отношении СД1 и уровня склеростина в литературе представлено несколько работ, подтверждающих его повышение у детей и взрослых с СД1 [19][23–25].
Отрицательная корреляция уровня склеростина с уровнем гликированного гемоглобина была описана Wedrychowicz A. у детей и подростков с СД1 [25]. Rubin M.R. также подтверждено значимое повышение склеростина при СД1, однако не выявили связи с уровнем гликированного гемоглобина [26]. Отличия в полученных результатах, вероятнее всего, обусловлены разницей в возрасте участников (дети в первом случае и взрослые во втором случае), так как уровень склеростина повышается в течение жизни, и более высокие его значения могли исказить взаимосвязь с HbA1c.
По результатам работы Neumann T и соавт. показатели склеростина у пациентов с СД1 были выше, чем в контрольной группе, однако авторы не отметили связи склеростина с маркерами метаболизма костной ткани. Таким образом, было выдвинуто предположение о том, что склеростин может быть связан с остеопорозом независимо от маркеров метаболизма костной ткани [19].
Отдельного внимания в рамках метаболического взаимодействия костной ткани и эндокринной функции поджелудочной железы заслуживает производное остеобластов — остеокальцин. В организме он существует в виде двух форм: карбоксилированная или «неактивная» форма остеокальцина, которая является важным строительным элементом костного матрикса, выступая в качестве цементирующего фактора при связывании коллагена с минерализованным компонентами кости, и декарбоксилированная или «активная форма» остеокальцина, которая образуется при воздействии остеокластов на костный матрикс и высвобождается в кровоток [27]. Активная форма остеокальцина оказывает влияние на клеточный метаболизм в качестве гормона. В поджелудочной железе остеокальцин воздействует на бета-клетки, стимулируя секрецию инсулина, и в то же время активирует продукцию адипонектина адипоцитами, который повышает чувствительность к инсулину. Таким образом, костная ткань через остеокальцин вносит вклад и в регуляцию гомеостаза глюкозы [2].
В 2007 г. Li и соавт. опубликовали результаты исследования наблюдения за животными с нокаутированным геном остеокальцина. Авторы исследовали мышей с избыточным весом и высоким уровнем глюкозы в крови при сниженном уровне инсулина. В результате эксперимента выявлено формирование инсулинорезистентности у мышей при выключении гена остеокальцина, что проявлялось в нарушении толерантности к глюкозе и снижении скорости инфузии глюкозы в исследованиях с гиперинсулинемическим эугликемическим клэмпом [28].
Данное открытие в сочетании с ранее известной ролью инсулина в стимулировании ремоделирования костной ткани выявило наличие прямой связи между костью и поджелудочной железой, взаиморегулирующими секрецию инсулина и метаболизм костной ткани [29].
В двух исследованиях на группах пациентов с СД2 (Xiao-Yu Ma и Jee-Aee Im) была продемонстрирована взаимосвязь уровня остеокальцина с нарушением углеводного обмена [30][31]. Значения остеокальцина у пациентов с СД были значительно ниже по сравнению с контрольной группой. Была выявлена отрицательная корреляция остеокальцина с показателями глюкозы и гликированного гемоглобина, а также корреляция с индексом HOMA.
Однако при анализе результатов проводилась оценка общего уровня остеокальцина без разделения на активную и неактивную форму, в то время как именно декарбоксилированная форма гормона оценивалась в исследованиях на мышах. Таким образом, остается открытым вопрос, какую роль играет декарбоксилированный остеокальцин в регуляции энергетического обмена у человека [32].
Характерной особенностью клеток при СД является снижение количества митохондрий и развитие митохондриальной дисфункции под влиянием хронической гипергликемии [33].
Дополнительное влияние на уровень энергообмена у пациентов с СД оказывают адипокины, такие как липокалин-2, также известный как нейтрофильный желатиназо-ассоциированный липокалин (NGAL). Имеются данные о выработке этого адипокина в костной ткани [34].
Mattia Capulli и соавт. продемонстрировали результаты удаления гена липокалина-2 на животной модели мышей. У мышей с инактивированным геном было выявлено снижение объема трабекулярной костной ткани и уменьшение остеобластов при отсутствии изменений остеокластов. При наличии у мышей повышенной массы тела и гиперфагии на фоне отсутствия липокалина-2 наблюдалась более низкая гликемия натощак, гиперинсулинемия, полиурия и глюкозурия. В совокупности эти результаты указывают на важную роль липокалина-2 в метаболизме костной ткани, подчеркивая связь с метаболизмом глюкозы [35].
Дальнейшие популяционные исследования W. Wang в Китае показали, что уровень липокалина-2 у пациентов с СД2 был значительно выше, чем в контрольной группе, и его экспрессия положительно коррелировала с уровнями воспалительных маркеров, таких как С-реактивный белок, интерлейкин-6 и фактор некроза опухолей-альфа [36].
Снижение скорости обновления костной ткани подтверждается регистрацией низких уровней маркеров костного обмена.
По результатам метаанализа K. Hygum и соавт. 2017 г. были выделены особенности изменения маркеров при СД. Снижение показателей С-терминального телопептида коллагена 1‑го типа, остеокальцина, N-терминального пропептида проколлагена 1‑го типа наблюдалось в группах пациентов с СД1 и СД2 независимо от типа СД по сравнению с группой контроля. При этом было отмечено повышение уровня склеростина в группах СД, однако при СД2 показатель был выше, чем в группе контроля и группе СД1. Тартрат-резистентная кислая фосфатаза была значительно ниже у пациентов с СД2 по сравнению с контролем. Уровень остеопротегерина был значительно выше при СД в целом по сравнению с контролем [37].
В физиологических условиях эндогенный инсулин приводит к увеличению выработки инсулиноподобного фактора роста 1 (ИФР-1), оказывая анаболическое действие на костные клетки. У пациентов с СД1 абсолютный дефицит инсулина приводит к снижению остеобластогенеза и обуславливает низкую скорость обновления костной ткани и снижение минеральной плотности кости [38].
M. Zhang и соавт. продемонстрировали дефекты минерализации и снижение плотности костной ткани при выключении гена, кодирующего рецептор ИФР-1 на примере мышей. Мыши, несущие эту специфичную для костной ткани мутацию, были нормального размера и веса, но по сравнению с нормальными мышами того же возраста у них наблюдалось значительное уменьшение объема губчатой костной ткани, количества трабекул, а также увеличение расстояния между трабекулами. При этом снижение скорости минерализации остеоида происходило при повышении активности остеобластов и остеокластов, которое фиксировалось во время эксперимента [39].
Несколько позже Tao Wang с соавт. представили результаты исследования по оценке влияния ИФР-1 на формирование энхондральной кости во время заживления переломов. Ученые оценили состояние костной мозоли через 10, 15, 21 и 28 дней после перелома большеберцовой кости на мышиной модели с нокаутом гена рецептора ИФР-1. При отсутствии влияния ИФР-1 наблюдался меньший размер костной мозоли с меньшим объемом кости, сопровождающийся дефектом минерализации и микроархитектурными аномалиями кости. Дополнительно было зафиксировано снижение маркеров костеообразования (остеокальцин, щелочная фосфатаза, коллаген 1α1) у мышей с выключенным ИФР-1, что подтверждало важную роль ИФР-1 в дифференцировке остеобластов во время заживления переломов [40].
Патогенетические механизмы костного ремоделирования у пациентов с СД2 в свою очередь связаны с изменением синтеза DPP-4 (dipeptidyl peptidase-4, дипептидилпептидаза-4) остеокластами и нарушением ранней дифференцировки остеобластов [41].
По результатам проспективного рандомизированного контролируемого исследования Megan M. Weivoda и соавт. были выделены специфические для остеокластов факторы, одним из которых стала DPP-4.
Под влиянием RANKL (Receptor activator of NF-κB ligand = Рецепторный активатор ядерного фактора каппа-Β лиганд) в остеокластах происходит экспрессия DPP-4, повышение которого способствует снижению уровня GLP-1 (глюкагоноподобного пептида-1), снижению инсулина и увеличению секреции глюкагона.
Взаимосвязь костного метаболизма с метаболизмом глюкозы через DPP-4 становится мишенью для терапевтического воздействия как со стороны антирезорбтивной терапии, так и со стороны сахароснижающих препаратов.
Авторы продемонстрировали значительное снижение циркулирующего DPP-4 и повышение GLP 1 в группе пациентов, получавших деносумаб, при сравнении с группой плацебо.
В своей второй работе с участием пациентов с СД2 и предиабетом авторы описали влияние терапии деносумабом на показатели углеводного обмена при сравнении с лечением препаратами бисфосфонатов, кальция и витамином D.
Исследования «случай-контроль» проводилось в клинике Мэйо в Рочестере с 2009 по 2016 гг. Включенные пациенты были в возрасте от 45 до 100 лет с остеопорозом, которые в течение как минимум 1 года принимали деносумаб, пероральные или внутривенные бисфосфонаты или добавки с кальцием и витамином D. 12-месячный курс лечения деносумабом снизил уровень HbA1c сильнее, чем прием бисфосфонатов или добавок с кальцием и витамином D [41].
Помимо влияния на маркеры костного метаболизма и скорость ремоделирования кости, СД способен оказывать воздействие на костный обмен со стороны жировой ткани.
Мезенхимальные стволовые клетки способны дифференцироваться не только в остеобласты, но и в адипоциты, в зависимости от чувствительности рецептора, активируемого пролифераторами пероксисом гамма-типа. При активации 2 изоформы рецептора клетки остеобластного происхождения превращаются в терминально дифференцированные адипоциты, нарушая баланс между адипогенезом и остеобластогенезом в костном мозге [42].
Изучение взаимосвязи жировой ткани с показателями костного обмена на животных моделях мышей с СД продемонстрировало отрицательную корреляцию между содержанием адипоцитов костного мозга и потерей трабекулярной и кортикальной кости [43].
Wei Li и соавт. продемонстрировали увеличение содержания жировой ткани в костном мозге большеберцовой кости у пациентов с впервые выявленным СД1 (n=35) при сравнении с контрольной группой. Авторы выделили обратную связь между долей жира в костном мозге и объемом и количеством трабекул костной ткани [44].
Vicente FC Andrade и соавт. описали влияние компенсации углеводного обмена при СД2 на количество адипоцитов в костном мозге. Процент адипоцитов преобладал в группе пациентов с уровнем гликированного гемоглобина более 7,0%. Пациенты с лучшим гликемическим контролем имели более низкие показатели процентного объема адипоцитов на объем костного мозга, а также качество кости [45].
Angela Sheu и соавт. предлагают выделять пациентов с СД как кандидатов с высоким риском переломов при наличии: уровня гликированного гемоглобина более 8.0%, длительности заболевания более 5 лет, наличия макро- и микрососудистых осложнений, а также при назначении пациентам инсулинотерапии, тиазолидиндионов, канаглифлозина [46].
В настоящее время не разработан идеальный алгоритм обследования и выявления остеопороза у пациентов с СД ввиду особенностей изменения костной ткани. Однако в клинической практике может быть использован ряд инструментов для повышения чувствительности методов диагностики: 1) выбор дополнительного фактора риска в виде ревматоидного артрита при наличии СД2 в рамках расчета риска основных остеопоротических переломов по FRAX (Fracture Risk Assessment=оценка риска переломов); 2) использование новой версии FRAX plus с возможностью выбора длительности СД; 3) определение трабекулярного костного индекса при проведении рентгеновской денситометрии, позволяющего оценивать косвенные показатели качества костной ткани; 4) оценка высоты тел грудных и поясничных позвонков лучевыми методами исследования с целью исключения компрессионных переломов у пациентов с некомпенсированным углеводным обменом или у пациентов на инсулинотерапии; 5) оценка минеральной плотности кости методом радиочастотной эхографической мультиспектрометрии (REMS) [46-49].
Понимание основных механизмов изменения метаболизма костной ткани при СД открывает возможности для оценки влияния новых сахароснижающих и других препаратов на процессы ремоделирования костной ткани в условиях хронической гипергликемии и возможной профилактики остеопоротических переломов [16][50].
Смещение равновесия костного ремоделирования в сторону усиления костной резорбции при возрастных изменениях отражает баланс между активацией и апоптозом остеокластов. Предполагается, что в физиологических условиях существует компенсаторный механизм, ограничивающий избыточную резорбцию кости, нарушение которого после менопаузы и в старшей возрастной группе способствует усиленной дифференцировке зрелых остеокластов и снижению их апоптоза.
В условиях остеопороза происходит формирование гипоксической среды, особенно в зонах резорбции, где уровень кислорода значительно ниже, чем в других участках тканей, что регулирует апоптоз остеокластов [12].
При гипоксии метаболическая адаптация остеокластов проявляется путем активации АМФ-активируемой протеинкиназы, что стимулирует аутофагию и усиливает дифференцировку остеокластов через подавление mTOR. Дополнительное влияние на активацию остеокластов при остеопорозе оказывает HIF-1. Исследования как in vivo, так и in vitro продемонстрировали повышение экспрессии HIF-1 и контролируемых им гликолитических ферментов (лактатдегидрогеназы, пируваткиназы, фосфофруктокиназы) в остеокластах при гипоксии в условиях остеопороза [12].
Помимо усиления гипоксии с течением возраста, у человека происходит нарушение целостности и функциональной активности митохондрий (рост повреждений митохондриальной ДНК, снижение количества белков дыхательной цепи, накопление активных форм кислорода), что приводит к формированию окислительного стресса и запуску катаболических процессов в клетках [2].
Кроме того, снижение эффективности репликации ДНК, транскрипции и синтеза аденозинтрифосфата (АТФ) при увеличении возраста негативно сказывается на костном ремоделировании: замедление обновления клеток препятствует обновлению костной ткани, а дефицит энергетического субстрата снижает ее прочность [2].
В развитии постменопаузального остеопороза важную роль играет воздействие окислительного стресса, ключевое значение при котором имеет нарушение метаболизма пуринов. Биосинтез пуриновых нуклеотидов осуществляется de novo из предшественников, включающих пентозофосфаты, аминокислоты, производные тетрагидрофолата и диоксид углерода, посредством многоступенчатого ферментативного каскада, и заканчивается катаболизм этих соединений ферментативным расщеплением до мочевой кислоты при помощи фермента ксантиноксидазы [51].
Ключевым фактором нарушения пуринового обмена выступает недостаточность фермента гипоксантин-гуанинфосфорибозилтрансферазы (ГГФТ). Физиологическая роль данного фермента заключается в реутилизации пуриновых оснований — он катализирует перенос фосфорибозильного остатка на гипоксантин и гуанин, превращая их, соответственно, в инозинмонофосфат (ИМФ) и гуанозинмонофосфат (ГМФ). Эти нуклеотиды, в свою очередь, выступают в роли естественных ингибиторов, регулирующих синтез пуринов de novo по принципу отрицательной обратной связи. При дефиците фермента продукция ИМФ и ГМФ снижается, что ослабляет ингибирующий контроль над биосинтезом пуринов. Следствием этого является компенсаторная активация синтеза пуринов de novo и значительное накопление мочевой кислоты — конечного продукта их катаболизма [51].
В физиологических концентрациях мочевая кислота оказывает протекторное действие на костную ткань, однако в условиях повышенной продукции оказывает повреждающее действие и способна увеличивать частоту остеопоротических переломов в группах риска, особенно среди женщин [52].
Увеличение образования мочевой кислоты подавляет систему антиоксидантной защиты и инициирует воспалительный ответ, что, в свою очередь, потенцирует костную резорбцию за счет активации остеокластов и ведет к потере костной массы. Параллельно реактивные формы кислорода индуцируют апоптоз остеобластов и стимулируют дифференцировку остеокластов, еще больше нарушая костный гомеостаз [53].
Накопление свободных радикалов способно индуцировать повреждение клеточных структур, включая генетический материал, а также нарушать фундаментальные метаболические процессы, такие как интенсификация перекисного окисления липидов и денатурация белковых молекул [51].
У женщин в постменопаузальном периоде регистрируется значительное повышение сывороточного уровня мочевой кислоты, демонстрирующее тесную корреляцию с ИМТ и количеством жировой ткани [51]. Данное явление отчасти объясняется ключевой ролью эстрогенов в пуриновом обмене. С одной стороны, эстрогены повышают биодоступность ключевых пуриновых производных, в частности циклического аденозинмонофосфата (цАМФ) [51], а экзогенная заместительная терапия эстрогенами ассоциирована с улучшением метаболизма пуринов и снижением риска развития гиперурикемии [54]. С другой стороны, элиминация пуриновых метаболитов также находится под гормональным контролем: экспрессия эстрогенового рецептора β в эпителии почечных канальцев необходима для регуляции выведения мочевой кислоты [55]. Таким образом, дефицит эстрогенов в постменопаузе способствует усиленному превращению пуринов в мочевую кислоту с последующим ее накоплением, что в совокупности с генерацией активных форм кислорода вносит существенный вклад в нарушение костного гомеостаза.
Патологическое усиление адипогенеза, наблюдаемое при ожирении, дополнительно подавляет остеогенную дифференцировку. Взаимосвязь ожирения с метаболизмом пуринов проявляется в нескольких аспектах: во-первых, жировая ткань является источником гипоксантина, концентрация которого в крови независимо коррелирует с выраженностью ожирения; во-вторых, при ожирении нарушается баланс ферментов ксантиндегидрогеназа/ксантиноксидаза (КО), что интенсифицирует катаболизм пуринов и продукцию активных форм кислорода (АФК) [51]. Таким образом, дисбаланс синтеза и распада пуринов играет важную роль в патогенезе остеопороза.
Кроме того, внеклеточный аденозинтрифосфат (АТФ) может повышать уровень внутриклеточного кальция и активировать вторичных посредников, что способствует нормальному функционированию остеобластов. Позже были получены данные, что остеобласты и остеокласты экспрессируют почти все рецепторы P2Y и P2X, опосредуя множественные клеточные реакции путем локальной и системной передачи сигналов для контроля обновления костной ткани [56].
Рецепторы этой системы классифицируются на P2- и P1-типы в зависимости от их лигандной специфичности: P2-рецепторы взаимодействуют преимущественно с нуклеотидами (например, АТФ), тогда как P1-рецепторы избирательно активируются нуклеозидом аденозином. Оба класса пуринергических рецепторов присутствуют на поверхности остеобластов и остеокластов, что подчеркивает их важность в костном метаболизме.
Аденозин, являющийся производным адениновых нуклеотидов (АТФ, АДФ, АМФ), в норме присутствует в организме в концентрациях 20–300 нМ [54], однако при развитии патологических процессов (травма, воспаление, клеточный стресс) наблюдается резкое, многократное повышение его внеклеточного уровня. Данное увеличение обусловлено стресс-индуцированной активацией ключевых ферментов эктонуклеотидазного каскада — эктонуклеозидтрифосфатдифосфогидролазы-1 (CD39) и экто-5′-нуклеотидазы (CD-73) [56]. В частности, при повреждении тканей внутриклеточный АТФ высвобождается через паннексиновые/коннексиновые каналы, после чего последовательно гидролизуется CD39 до АМФ, а затем CD73 до аденозина. Помимо этого, тканенеспецифическая щелочная фосфатаза (TNAP) также вносит вклад в генерацию внеклеточного аденозина, катализируя прямое расщепление АТФ [56].
Воздействие на аденозиновые рецепторы путем их активации или ингибирования оказывает существенное влияние на гомеостаз костной ткани. Помимо регуляции дифференцировки остеобластов и остеокластов, внеклеточный аденозин выполняет иммуномодулирующую функцию через привлечение нейтрофилов и подавление Т-клеток, одновременно стимулируя иммуносупрессивные клетки, что играет роль в разрешении воспаления в процессе заживления костей скелета.
Недавно было продемонстрировано снижение у животных с овариоэктомией внеклеточного аденозина в костном мозге, а также снижение экспрессии CD-39 и CD-73, что свидетельствовало о сниженной сигнализации аденозина в условиях постменопаузы и остеопороза [57]. В свою очередь применение молекул, повышающих активность внеклеточного аденозина (таких как дипиридамол), предотвращало снижение костной массы у животных после удаления яичников [56].
Терапевтический потенциал представляет использование лекарственных препаратов, направленных на модуляцию аденозин-опосредованных сигнальных путей в процессе репарации костной ткани. Однако современная фармакология сталкивается с ограничениями, такими как быстрый полураспад аденозина (менее 10 секунд), низкая биодоступность, а также развитие побочных эффектов при его введении со стороны сердечно-сосудистой системы, в связи с чем продолжается поиск оптимального носителя для доставки аденозина в кость [60].
Удачное применение алендроновой кислоты в качестве носителя аденозина описано в эксперименте лечения остеопороза у мышей. При комбинированном действии лекарственная терапия позволяла не только снижать потерю костной массы, но и оказывала увеличение образования костной ткани, поскольку аденозин не только ингибировал остеокластогенез, но и стимулировал линию остеобластогенеза [59].
Проведенный анализ демонстрирует центральную роль энергетического метаболизма в регуляции костного ремоделирования. Костные клетки обладают метаболической пластичностью, динамически переключаясь между гликолизом и окислительным фосфорилированием на разных этапах дифференцировки. Ключевые сигнальные пути (HIF, Wnt/β-катенин, AMPK/mTOR) интегрируют метаболические и морфогенетические сигналы, обеспечивая адаптацию биоэнергетики к функциональным потребностям клеток. В условиях гипергликемии изменение метаболизма обусловлено накоплением конечных продуктов гликирования, повышением уровня адипокинов, а также снижением скорости ремоделирования костной ткани. Ключевыми патогенетическими механизмами при остеопорозе выступают нарушение пуринового обмена с накоплением мочевой кислоты, развитие окислительного стресса и гипоксии костной ткани, а также возрастное снижение активности митохондрий. Существенный вклад в патогенез вносят нарушения пуринового обмена и дисрегуляция аденозиновой сигнализации. Изучение нарушений энергообмена при патологии скелета, особенно в условиях нарушения углеводного обмена, в частности СД, представляет интерес для полноценного понимания звеньев патогенеза и разработки таргетных препаратов для профилактики и лечения первичного и вторичного остеопороза и ассоциированных с ним патологических переломов.
Источники финансирования. Работа выполнена в рамках государственного задания №124020700097-8.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Белая Ж.Е. — концепция, внесение в рукопись существенной правки, одобрение финальной версии рукописи; Жданова А.С. — анализ данных, написание статьи; Катаева Д.А. — концепция и дизайн статьи; Омельченко К.А. — внесение в рукопись правок, одобрение финальной версии рукописи. Все авторы одобрили финальную версию статьи.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
1. Srivastava RK, Sapra L, Mishra PK. Osteometabolism: Metabolic Alterations in Bone Pathologies. Cells. 2022;11(23):3943. doi: https://doi.org/10.3390/cells11233943
2. Sautchuk RJr, Eliseev RA. Cell energy metabolism and bone formation. Bone Rep. 2022;16:101594. doi: https://doi.org/10.1016/j.bonr.2022.101594
3. Wu H, Yin G, Pu X, Wang J, Liao X, Huang Z. Coordination of Osteoblastogenesis and Osteoclastogenesis by the Bone Marrow Mesenchymal Stem Cell-Derived Extracellular Matrix To Promote Bone Regeneration. ACS Appl Bio Mater. 2022;5(6):2913–2927. doi: https://doi.org/10.1021/acsabm.2c00264
4. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. 2021;599(1):23–37. doi: https://doi.org/10.1113/JP280572
5. Гребенникова Т.А., Белая Ж.Е., Рожинская Л.Я., Мельниченко Г.А. Канонический сигнальный путь Wnt/β-катенин: от истории открытия до клинического применения // Терапевтический архив. — 2016. — Т. 88. — №10. — С. 74–81. doi: https://doi.org/10.17116/terarkh201688674-81
6. Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. 2019;20(7):436–450. doi: https://doi.org/10.1038/s41580-019-0123-5
7. Smith CO, Eliseev RA. Energy Metabolism During Osteogenic Differentiation: The Role of Akt. Stem Cells Dev. 2021;30(3):149–162. doi: https://doi.org/10.1089/scd.2020.0141
8. Karner CM, Lee SY, Long F. Bmp Induces Osteoblast Differentiation through both Smad4 and mTORC1 Signaling. Mol Cell Biol. 2017;37(4):e00253–16. doi: https://doi.org/10.1128/MCB.00253-16
9. Huang F, Hu L, Zhang Y, Qu X, Xu J. BMP4 Moderates Glycolysis and Regulates Activation and Interferon-Gamma Production in CD4+ T Cells. Front Immunol. 2021;12:702211. doi: https://doi.org/10.3389/fimmu.2021.702211
10. Li Y, Su J, Sun W, Cai L, Deng Z. AMP-activated protein kinase stimulates osteoblast differentiation and mineralization through autophagy induction. Int J Mol Med. 2018;41(5):2535–2544. doi: https://doi.org/10.3892/ijmm.2018.3498
11. Fitter S, Matthews MP, Martin SK, et al. mTORC1 Plays an Important Role in Skeletal Development by Controlling Preosteoblast Differentiation. Mol Cell Biol. 2017;37(7):e00668–16. doi: https://doi.org/10.1128/MCB.00668-16
12. Da W, Tao L, Zhu Y. The Role of Osteoclast Energy Metabolism in the Occurrence and Development of Osteoporosis. Front Endocrinol (Lausanne). 2021;12:675385. doi: https://doi.org/10.3389/fendo.2021.675385
13. Taubmann J, Krishnacoumar B, Böhm C, et al. Metabolic reprogramming of osteoclasts represents a therapeutic target during the treatment of osteoporosis. Sci Rep. 2020;10(1):21020. doi: https://doi.org/10.1038/s41598-020-77892-4
14. Morten KJ, Badder L, Knowles HJ. Differential regulation of HIF-mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts. J Pathol. 2013;229(5):755–764. doi: https://doi.org/10.1002/path.4159
15. Ялочкина Т.О., Белая Ж.Е. Низкотравматичные переломы и костное ремоделирование при сахарном диабете 2 типа // Ожирение и метаболизм. — 2017. — Т. 14. — №3. — С. 11–18. doi: https://doi.org/10.14341/omet2017311-18
16. Каронова Т.Л., Тимкина Н.В., Радугин Ф.М., Симаненкова А.В., Черникова А.Т., Гринева Е.Н. Особенности костного ремоделирования при сахарном диабете. // Остеопороз и остеопатии. — 2022. — Т.25. — №3. — С.63-64. [Karonova TL, Timkina NV, Radugin FM, et al. Osobennosti kostnogo remodelirovaniya pri sakharnom diabete. Osteoporosis and Bone Diseases. 2022;25(3):63-64. (In Russ.) doi: https://doi.org/10.14341/osteo13031
17. Farlay D, Armas LA, Gineyts E, Akhter MP, Recker RR, Boivin G. Nonenzymatic Glycation and Degree of Mineralization Are Higher in Bone From Fractured Patients With Type 1 Diabetes Mellitus. J Bone Miner Res. 2016;31(1):190–195. doi: https://doi.org/10.1002/jbmr.2607
18. Neumann T, Lodes S, Kästner B, et al. High serum pentosidine but not esRAGE is associated with prevalent fractures in type 1 diabetes independent of bone mineral density and glycaemic control. Osteoporos Int. 2014;25(5):1527–1533. doi: https://doi.org/10.1007/s00198-014-2631-7
19. Neumann T, Hofbauer LC, Rauner M, et al. Clinical and endocrine correlates of circulating sclerostin levels in patients with type 1 diabetes mellitus. Clin Endocrinol (Oxf). 2014;80(5):649–655. doi: https://doi.org/10.1111/cen.12364
20. Gaudio A, Privitera F, Battaglia K, et al. Sclerostin levels associated with inhibition of the Wnt/β-catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(10):3744–3750. doi: https://doi.org/10.1210/jc.2012-1901
21. Гребенникова Т.А., Белая Ж.Е., Рожинская Л.Я., и др. Эпигенетические аспекты остеопороза // Вестник Российской академии медицинских наук. — 2015. — Т. 70. — №5. — С. 541–548. doi: https://doi.org/10.15690/vramn.v70.i5.1440
22. García-Martín A, Rozas-Moreno P, Reyes-García R, et al. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(1):234–241. doi: https://doi.org/10.1210/jc.2011-2186
23. Kurban S, Selver Eklioglu B, Selver MB. Investigation of the relationship between serum sclerostin and dickkopf-1 protein levels with bone turnover in children and adolescents with type-1 diabetes mellitus. J Pediatr Endocrinol Metab. 2022;35(5):673–679. doi: https://doi.org/10.1515/jpem-2022-0001
24. Gennari L, Merlotti D, Valenti R, et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J Clin Endocrinol Metab. 2012;97(5):1737–1744. doi: https://doi.org/10.1210/jc.2011-2958
25. Wędrychowicz A, Sztefko K, Starzyk JB. Sclerostin and its significance for children and adolescents with type 1 diabetes mellitus (T1D). Bone. 2019;120:387–392. doi: https://doi.org/10.1016/j.bone.2018.08.007
26. Rubin MR, de Boer IH, Backlund JC, et al. Biochemical Markers of Bone Turnover in Older Adults With Type 1 Diabetes. J Clin Endocrinol Metab. 2022;107(6):e2405–e2416. doi: https://doi.org/10.1210/clinem/dgac099
27. Moser SC, van der Eerden BCJ. Osteocalcin-A Versatile Bone-Derived Hormone. Front Endocrinol (Lausanne). 2019;9:794. doi: https://doi.org/10.3389/fendo.2018.00794
28. Lee NK, Sowa H, Hinoi E, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi: https://doi.org/10.1016/j.cell.2007.05.047
29. Motyl KJ, Guntur AR, Carvalho AL, Rosen CJ. Energy Metabolism of Bone. Toxicol Pathol. 2017;45(7):887–893. doi: https://doi.org/10.1177/0192623317737065
30. Ma XY, Chen FQ, Hong H, Lv XJ, Dong M, Wang QY. The Relationship between Serum Osteocalcin Concentration and Glucose and Lipid Metabolism in Patients with Type 2 Diabetes Mellitus – The Role of Osteocalcin in Energy Metabolism. Ann Nutr Metab. 2015;66(2-3):110–116. doi: https://doi.org/10.1159/000370198
31. Im JA, Yu BP, Jeon JY, Kim SH. Relationship between osteocalcin and glucose metabolism in postmenopausal women. Clin Chim Acta. 2008;396(1-2):66–69. doi: https://doi.org/10.1016/j.cca.2008.07.001
32. Гребенникова Т.А., Белая Ж.Е., Цориев Т.Т., и др. Эндокринная функция костной ткани // Остеопороз и остеопатии. — 2015. — Т. 18. — №1. — С. 28–37. doi: https://doi.org/10.14341/osteo2015128-37
33. Liu H, Wang S, Wang J, et al. Energy metabolism in health and diseases. Signal Transduct Target Ther. 2025;10(1):69. doi: https://doi.org/10.1038/s41392-025-02141-x
34. Yang Y, Liu J, Kousteni S. Lipocalin 2-A bone-derived anorexigenic and β-cell promoting signal: From mice to humans. J Diabetes. 2024;16(3):e13504. doi: https://doi.org/10.1111/1753-0407.13504
35. Capulli M, Ponzetti M, Maurizi A, et al. A Complex Role for Lipocalin 2 in Bone Metabolism: Global Ablation in Mice Induces Osteopenia Caused by an Altered Energy Metabolism. J Bone Miner Res. 2018;33(6):1141–1153. doi: https://doi.org/10.1002/jbmr.3406
36. Wang W, Ye S, Qian L, et al. Elevated serum lipocalin 2 levels are associated with indexes of both glucose and bone metabolism in type 2 diabetes mellitus. Endokrynol Pol. 2018;69(3):276–282. doi: https://doi.org/10.5603/EP.a2018.0030
37. Hygum K, Starup-Linde J, Harsløf T, et al. MECHANISMS IN ENDOCRINOLOGY: Diabetes mellitus, a state of low bone turnover - a systematic review and meta-analysis. Eur J Endocrinol. 2017;176(3):R137–R157. doi: https://doi.org/10.1530/EJE-16-0652
38. Демидова Т.Ю., Плахотняя В.М. Сахарный диабет и остеопороз: патогенетическая связь и современные принципы лечения // Медицинский совет. — 2021. — №7. — С. 96–107doi: https://doi.org/10.21518/2079-701X-2021-7-96-107
39. Zhang M, Xuan S, Bouxsein ML, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277(46):44005–44012. doi: https://doi.org/10.1074/jbc.M208265200
40. Wang T, Wang Y, Menendez A, et al. Osteoblast-Specific Loss of IGF1R Signaling Results in Impaired Endochondral Bone Formation During Fracture Healing. J Bone Miner Res. 2015;30(9):1572-1584. doi: https://doi.org/10.1002/jbmr.2510
41. Weivoda MM, Chew CK, Monroe DG, et al. Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat Commun. 2020;11(1):87. doi: https://doi.org/10.1038/s41467-019-14003-6
42. Sanches CP, Vianna AGD, Barreto FC. The impact of type 2 diabetes on bone metabolism. Diabetol Metab Syndr. 2017;9:85. doi: https://doi.org/10.1186/s13098-017-0278-1
43. Kim TY, Schafer AL. Diabetes and Bone Marrow Adiposity. Curr Osteoporos Rep. 2016;14(6):337–344. doi: https://doi.org/10.1007/s11914-016-0336-x
44. Li W, Wang W, Zhang M, et al. Associations of marrow fat fraction with MR imaging based trabecular bone microarchitecture in first-time diagnosed type 1 diabetes mellitus. Front Endocrinol (Lausanne). 2024;15:1287591. doi: https://doi.org/10.3389/fendo.2024.1287591
45. Andrade VFC, Besen D, Chula DC, et al. Bone Marrow Adiposity in Premenopausal Women With Type 2 Diabetes With Observations on Peri-Trabecular Adipocytes. J Clin Endocrinol Metab. 2021;106(9):e3592–e3602. doi: https://doi.org/10.1210/clinem/dgab322
46. Sheu A, White CP, Center JR. Bone metabolism in diabetes: a clinician's guide to understanding the bone-glucose interplay. Diabetologia. 2024;67(8):1493–1506. doi: https://doi.org/10.1007/s00125-024-06172-x
47. Leslie WD, Johansson H, McCloskey EV, et al. Comparison of Methods for Improving Fracture Risk Assessment in Diabetes: The Manitoba BMD Registry. J Bone Miner Res. 2018;33(11):1923-1930. doi: https://doi.org/10.1002/jbmr.3538
48. Белая Ж.Е., Белова К.Ю., Бирюкова Е.В., и др. Федеральные клинические рекомендации по диагностике, лечению и профилактике остеопороза // Остеопороз и остеопатии. — 2021. — Т. 24. — №2. — С. 4–47. doi: https://doi.org/10.14341/osteo12930
49. Caffarelli C, Tomai Pitinca MD, Al Refaie A, et al. Ability of radiofrequency echographic multispectrometry to identify osteoporosis status in elderly women with type 2 diabetes. Aging Clin Exp Res. 2022;34(1):121–127. doi: https://doi.org/10.1007/s40520-021-01889-w
50. Радугин Ф.М., Каронова Т.Л. Особенности костного ремоделирования у больных сахарным диабетом: фокус на витамин К2 // Остеопороз и остеопатии. — 2021. — Т. 24. — №3. — С. 11–18. doi: https://doi.org/10.14341/osteo12929
51. Yang K, Li J, Tao L. Purine metabolism in the development of osteoporosis. Biomed Pharmacother. 2022;155:113784. doi: https://doi.org/10.1016/j.biopha.2022.113784
52. Terán D, Doleželová E, Keough DT, et al. Crystal structures of Trypanosoma brucei hypoxanthine - guanine - xanthine phosphoribosyltransferase in complex with IMP, GMP and XMP. FEBS J. 2019;286(23):4721–4736. doi: https://doi.org/10.1111/febs.14987
53. Lin KM, Lu CL, Hung KC, et al. The Paradoxical Role of Uric Acid in Osteoporosis. Nutrients. 2019;11(9):2111. doi: https://doi.org/10.3390/nu11092111
54. He H, Pan L, Liu F, et al. The Mediation Effect of Body Composition on the Association Between Menopause and Hyperuricemia: Evidence From China National Health Survey. Front Endocrinol (Lausanne). 2022;13:879384. doi: https://doi.org/10.3389/fendo.2022.879384
55. Liu H, Peng L, Ma J, et al. Low expression of estrogen receptor β in renal tubular epithelial cells may cause hyperuricemia in premenopausal patients with systemic lupus erythematosus. Lupus. 2021;30(4):560–567. doi: https://doi.org/10.1177/0961203320984231
56. Newman H, Varghese S. Extracellular adenosine signaling in bone health and disease. Curr Opin Pharmacol. 2023;70:102378. doi: https://doi.org/10.1016/j.coph.2023.102378
57. Shih YV, Liu M, Kwon SK, et al. Dysregulation of ectonucleotidase-mediated extracellular adenosine during postmenopausal bone loss. Sci Adv. 2019;5(8):eaax1387. doi: https://doi.org/10.1126/sciadv.aax1387
58. Albrecht-Küpper BE, Leineweber K, Nell PG. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal. 2012;8(Suppl 1):91–99. doi: https://doi.org/10.1007/s11302-011-9274-3
59. Hoque J, Shih YV, Zeng Y, et al. Bone targeting nanocarrier-assisted delivery of adenosine to combat osteoporotic bone loss. Biomaterials. 2021;273:120819. doi: https://doi.org/10.1016/j.biomaterials.2021.120819
Жданова Анастасия Станиславовна – аспирант.
117292, Москва, ул. Дмитрия Ульянова, д. 11
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Белая Жанна Евгеньевна - д.м.н.
Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Катаева Дарья Альбертовна - клинический ординатор.
Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
Омельченко Константин Анатольевич - к.м.н.
Москва
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи
|
|
1. Рисунок 1. Биоэнергетическая регуляция и сигнальные пути в процессе дифференцировки остеобластов (создано при помощи BioRender). | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(879KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Особенности энергетического обмена в остеокласте в процессе остеокластогенеза (создано при помощи BioRender). | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(834KB)
|
Метаданные ▾ | |
|
|
3. Рисунок 3. Общие и отличные патогенетические механизмы нарушения костного ремоделирования при сахарном диабете 1 и 2 типа. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(787KB)
|
Метаданные ▾ | |
Жданова А.С., Белая Ж.Е., Катаева Д.А., Омельченко К.А. Изменения энергетического обмена костной ткани при сахарном диабете и старении как причина повышенной хрупкости скелета. Сахарный диабет. 2026;29(2):191-202. https://doi.org/10.14341/DM13416
Zhdanova A.S., Belaya Zh.E., Kataeva D.A., Omelchenko K.A. Changes in bone tissue energy metabolism in diabetes mellitus and aging as a cause of increased skeletal fragility. Diabetes mellitus. 2026;29(2):191-202. (In Russ.) https://doi.org/10.14341/DM13416

 |
Адрес: 117036, Российская Федерация, Москва, улица Дмитрия Ульянова, дом 11.
Обработка персональных данных







































